ХОЛЕСТЕРИН
Холестерин (ХС) — стероид, характерный только для животных организмов.
Источником ХС в организме являются синтетические процессы и пища. В сутки в организме синтезируется около 1г (0.7) ХС. В печени синтезируется более 50% ХС, в тонком кишечнике — 15— 20%, остальной ХС синтезируется в коже, коре надпочечников, половых железах. С пищей поступает в сутки 0,3—0,5г (0.3-0.4) ХС. Общее содержание ХС в организме составляет в среднем 140г, 90-93% находиться в клетках, 7-10% - в крови (5,2+1,3 ммоль/л).
Биологическая роль ХС
1. ХС входит в состав всех мембран клеток, увеличивает их электроизоляционные свойства, придает им жесткость и прочность;
2. В мембране ХС защищает полиненасыщенные ЖК от окисления;
3. из ХС синтезируются жёлчные кислоты (0,5-0,7 г ХС в сут) 0.45, стероидных гормоны (половые и кортикоиды) (40 мг ХС в сут) и витамин Д3 (10 мг ХС в сут).
4. ХС является компонентом желчи, участвует в переваривании липидов.
Обмен ХС чрезвычайно сложен, в нем участвует около 300 разных белков.
Синтез ХС
Реакции синтеза ХС происходят в цитозоле и ЭПР клеток. Это один из самых длинных метаболических путей в организме человека (около 100 последовательных реакций).
Синтез ХС делят на 3 этапа:
I этап синтеза ХС - образование мевалоната (мевалоновой кислоты).
1. Две молекулы ацетил-КоА конденсируются тиолазой с образованием ацетоацетил-КоА;
2. Гидроксиметилглутарил-КоА-синтаза присоединяет третий ацетильный остаток к ацетоацетил-КоА с образованием ГМГ-КоА (3-гидрокси-3-метилглутарил-КоА). Эта последовательность реакций сходна с начальными стадиями синтеза КТ. Однако синтез КТ происходит в митохондриях печени, а реакции синтеза ХС — в цитозоле клеток.
3. ГМГ-КоА-редуктаза восстанавливает ГМГ-КоА до мевалоната с использованием 2 молекул НАДФH2. Фермент ГМГ-КоА-редуктаза — гликопротеин, пронизывающий мембрану ЭПР, активный центр которого выступает в цитозоль.
II этап синтеза ХС - образование сквалена
1. Мевалонат превращается в изопреноидную структуру — изопентенилпирофосфат (5 атомов С);
2. 2 изопентенилпирофосфата конденсируются в геранилпирофосфат (10 атомов С);
3. Присоединение изопентенилпирофосфата к геранилпирофосфату дает фарнезилпирофосфат (15 атомов С).
4. 2 фарнезилпирофосфата конденсируются в сквален (15 атомов С).
III этап синтеза ХС - образование ХС
Сквален циклазой превращается в ланостерол, (4 цикла и 30 атомов С).
Далее происходит 20 последовательных реакций, превращающих ланостерол в ХС (27 атомов С).
В организме человека изопентенилпирофосфат также служит предшественником убихинона (KoQ) и долихола, участвующего в синтезе гликопротеинов.
Регуляция синтеза ХС
Ключевой фермент синтеза ХС ГМГ-КоА-редуктаза регулируется несколькими способами:
· ХС, желчные кислоты (в печени) репрессируют ген ГМГ-КоА-редуктазы. В норме поступление ХС с пищей снижает синтез собственного ХС в печени, однако с возрастом эффективность этой регуляции у многих людей снижается и уровень ХС повышается.
· Инсулин через дефосфорилирование осуществляет активацию ГМГ-КоА-редуктазы.
· Глюкагон через фосфорилирование осуществляет ингибирование ГМГ-КоА-редуктазы.
Повышение концентрации исходного субстрата ацетил-КоА стимулирует синтез ХС.
Таким образом, синтез ХС активируется при питании углеводами и ингибируется при голодании.
Этерификация ХС
ХС образует с ЖК сложные эфиры (ЭХС), которые более гидрофобны чем сам ХС. В клетках эту реакцию катализирует АХАТ (ацилКоА: холестеролацилтрансферазой): ХС + АцилКоА → ЭХС + HSKoA
АХАТ содержится лишь в некоторых тканях, синтезированный им ЭХС формирует в цитоплазме липидные капли, которые являются формой хранения ХС. По мере необходимости ЭХС гидролизуются холестеролэстеразой на ХС и ЖК.
ЭХС синтезируются также в крови в ЛПВП под действием ЛХАТ (лецетин: холестеролацилтрансферазой): ХС + лецитин → ЭХС + лизолецитин
В составе ЛП ЭХС обеспечивают большую часть транспорта ХС в крови. На долю ЭХС крови приходиться 75% от общего количества ЭХС в организме.
Выведение ХС из организма
Так как производные циклопентанпергидрофенантрена (стероиды) водонерастворимы и в организме не расщепляются, они выводятся из организма в основном с калом в составе желчи и немного с потом через кожу.
В сутки из организма выводится от 1,0г до 1,3г ХС. ХС выводится с желчью (0,5-0,7 г/сут) в основном в виде жёлчных кислот и частично в чистом виде. Часть ХС в кишечнике под действием ферментов бактерий восстанавливается по двойной связи, образуя холестанол и копростанол. С кожным салом в сутки выделяется 0,1г ХС.
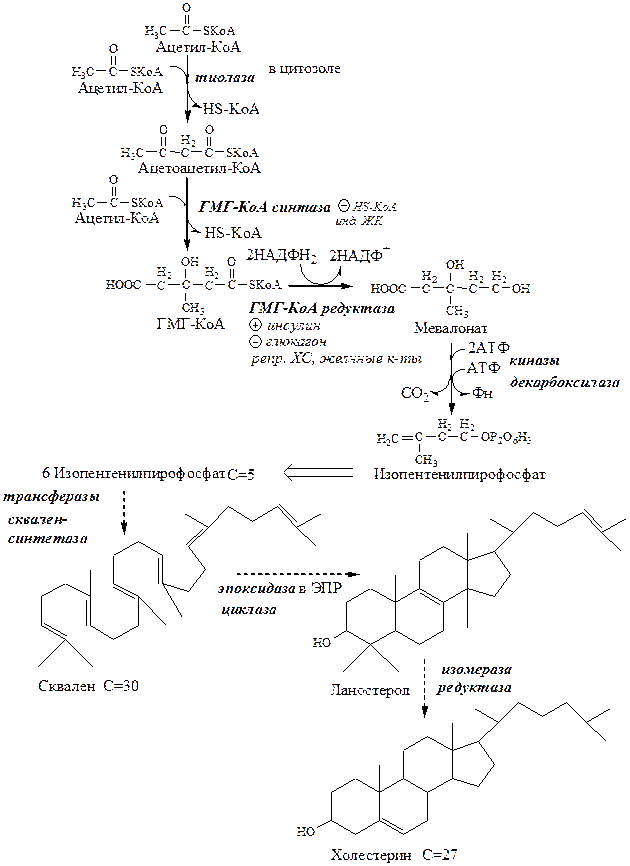
ГИПЕРХОЛЕСТЕРОЛЕМИЯ
Концентрация ХС в крови взрослых составляет 5,2+1,2 ммоль/л, как правило, с возрастом она увеличивается. Нарушения обмена ХС чаще всего проявляется гиперхолестеролемией, повышением ХС в крови выше нормы.
Причины развития гиперхолестеринемии:
1. Избыточного поступления с пищей ХС. Так как выведение из организма ХС ограничено 1,2—1,5 г/сут, излишки ХС накапливаются;
2. Переедание, недостаточная физическая активность, ожирение, сахарный диабет и гипотериоз способствуют гипергликемии и гиперлипидемии. Избыток углеводов и липидов в организме идет на повышенный синтез ХС;
3. Избыток в пище насыщенных и дефицит полиненасыщенных ЖК стимулирует в организме синтез ХС;
4. Некоторые дислипопротеинемии. Любой дефект рецептора ЛПНП (часто) или белка апоВ-100, взаимодействующего с ним, приводит к распространённому наследственному заболеванию — семейной гиперхолестеролемии. Она сопровождается ксантоматозом и атеросклерозом. У гомозигот с дефектом рецептора ЛПНП смерть в возрасте 5—6 лет от инфаркта или инсульта;
Ещё в 1987 г. Goldstein и Brown окончательно установили, что причиной семейной гиперхолестеролемии (СГХ) является дефект гена, ответственного за синтез апоВ-100 ЛПНП-рецептора на мембране гепатоцитов. У больных гетерозигот количество рецепторов снижено на 50%, а уровень ЛПНП крови повышен вдвое, что сочетается с ускоренным развитием атеросклероза и ишемической болезни сердца с инфарктом миокарда уже в юном возрасте.
Коэффициент атерогенности = (ХСобщ –ХСЛПВП) / ХСЛПВП < 3
Гиперхолестеринемия вызывает атеросклероз и желчекаменную болезнь.
Статины – «золотой стандарт» терапии гиперхолестеринемии и атеросклероза – наиболее широко применяемая сейчас группа препаратов, доказавшая свою эффективность при ИБС и других формах атеросклероза во многих клинических исследованиях [Мартынов АИ и соавт., 1997; Кухарчук ВВ и соавт., 2003; Шевченко ОП, 2003]. Однако, было показано, что эти препараты оказывают некоторый токсический эффект на печень, пищеварительную систему и мышечную ткань. Кроме того, лечение статинами пожизненное, поскольку при прекращении их приёма наблюдается выраженный синдром отмены. Достаточно широкое применение статинов ограничивает и их высокая стоимость [Преображенский ДВ и соавт., 1995; Шевченко ОП, 2003]. Широкому применению других гиполипидемических препаратов препятствуют высокая частота побочных эффектов, риск возникновения гепатотоксичности и других органотоксических эффектов (фибраты, никотиновая кислота, анионообменные смолы) или менее выраженный эффект (пробукол и др.) [Джанашия ПХ, 1998; Аронов ДМ, 2000; Робинс Д, 2001].
Для лечения некоторых категорий больных атеросклерозом (например семейной гиперхолестеринемией), резистентных к диетической и гиполипидемической терапии, применяется ЛПНП-аферез. Метод заключается в экстракорпоральной сорбции из крови апоВ-содержащих ЛП с помощью специальных иммуносорбентов или декстранцеллюлозы. Эффект процедуры является значительным (снижение концентрации ХС (ЛПНП) до 80%), но кратковременным. Необходимы повторные пожизненные сеансы как минимум 1 раз в месяц [Малышев ПП, 1997; Чебышев АН, 2000; Bambauer R., 2002]. В связи со сложностью и высокой стоимостью данного способа лечения он может применяться у весьма ограниченного круга больных.
Инвазивные хирургические методы применяются лишь для лечения осложнений атеросклероза [Савельев ВС и соавт., 1999] и так же исключают системный подход терапии.
Показано, что при гиперхолестеринемии и атеросклерозе наблюдается выраженная дисфункция сосудистого эндотелия – нарушение его вазодилатирующей способности [137, 104, 166]. Нарушение эндотелийзависимого расслабления сосудов обусловлено изменением метаболизма оксида азота (NO) в стенке сосуда (изменение активности или дефицит субстрата для NO-синтазы, ускоренная деструкция NO и т.д.) [14, 82, 85,126,130]. Основным фактором, повреждающим эндотелий при ДЛП, являются модифицированные ЛПНП. Исследование дисфункции эндотелия в ходе атерогенеза стимулировало изучение микроциркуляции в эксперименте и клинике.
Дислипидемии и дисфункция эндотелия обусловливают возникновение дислипидогенной микроангиопатии [17,139].
Ранее было установлено, что дислипидогенная микроангиопатия является инициальным звеном в возникновении и прогрессировании хронической неспецифической органной патологии и атеросклеротических изменений в органных и магистральных артериях у кроликов [17,35,54]. Показано, что такие изменения микроциркуляции как спазм артериол и дилатация венул, эритроцитарные микростазы возникают с первых часов и дней АТД, то есть на самых ранних стадиях дислипопротеидемии. При прогрессировании ДЛП и атерогенеза наблюдается образование эритроцитарных агрегатов по типу «монетных столбиков» или более плотных, характерных для сладж-синдрома, капилляротромбоз, разрежение и выключение из кровотока капиллярных сетей. Появляются внесосудистые изменения в виде диапедеза эритроцитов, клеточных инфильтратов и периваскулярного отёка [17,16,19].
В последние годы появились работы, свидетельствующие о ведущей роли расстройств микроциркуляции при ишемической болезни сердца и других формах сосудистой патологии у человека. Описаны отдельные формы ИБС, обусловленные первичной дисфункцией микроциркуляторной системы сердца [123,129]. На клиническом материале было показано, что ИБС может быть обусловлена не столько нарушением перфузии миокарда через атеросклеротические коронарные артерии, сколько нарушением микроциркуляции сердца [40,114]. В настоящее время предложены методы диагностики (ультразвуковая доплерография) и коррекции (плазмаферез, ультрафиолетовое облучение крови) микроциркуляторных нарушений при атеросклерозе [1].
Таким образом, дислипидогенная микроангиопатия является первичной генерализованной реакцией эндотелия на гиперлипопротеидемию и приводит к циркуляторной гипоксии и полиорганной патологии (дистрофии) ещё до появления атеросклеротических бляшек в органных и магистральных артериях.
1.1.3 Дисфункция иммунорегуляторной системы как фактор развития ДЛП и атеросклероза.
Гомеостатическая функция иммунорегуляторной системы в организме осуществляется с помощью двух ветвей: системы мононуклеарных фагоцитов, образуемой клетками макрофагально-моноцитарного ряда, и системы лимфоидной ткани, образуемой различными популяциями лимфоцитов.
Клетки макрофагально-моноцитарного ряда осуществляют первую - воспалительную линию защиты организма. В ответ на воспалительную реакцию лимфоидная ткань запускает восстановительные процессы, осуществляемые с помощью двух важнейших функций лимфоцитов: иммунорегуляторной, обеспечивающей антителогенез, и морфорегуляторной, обеспечивающей постоянство численного состава клеток (паренхиматозных органов), их структурный и функциональный гомеостаз [4].
Очевидно, развивающаяся на фоне ДЛП и атеросклероза дисрегуляция иммуннорегуляторной системы с гиперсекрецией острофазных белков, приводит к угнетению лимфоидной ткани и подавлению восстановительных процессов в повреждёной печени и других органах. Это создаёт условия для преобладающего воздействия на них системной воспалительной реакции, которая не будучи сбалансирована, выступает в роли фактора прогрессирования атеросклеротического процесса.
Клетки rупфера, представляя собой клетки макрофагально-моноцитарного ряда, и являясь ключевыми эффекторами воспаления в организме, оказывают в тоже время регуляторное воздействие на гепатоциты, модулируя их функциональную активность [38,39]. Эндотелиоциты печени также вырабатывают медиаторы воспаления и иммунной защиты [3], участвуя, таким образом, в осуществлении эффекторных реакций печени и всего организма.
Одной из таких реакций – является способность гепатоцитов продуцировать острофазные белки: С-реактивный белок, сывороточный амилоид А, гаптоглобин и др., которым принадлежит важная роль в инициации и модулировании воспалительных и репаративных ответов организма [3]. Так, например, показано, что при содержании кроликов на ХС-диете уже на 4-7 дни параллельно с развитием гиперхолестеринемии происходило резкое увеличение концентрации СРБ в крови. При этом установлено, что острофазная реакция гепатоцита предшествует его жировой дистрофии [32].
Предполагается, что острофазные белки не только манифестируют, но провоцируют развитие ДЛП и атеросклероза. Показано [31], что острофазные белки блокируют апоВ-100 рецепторный эндоцитоз ЛПНП, легко и прочно связываются с ЛП и становятся физиологическими аналогами их апобелков. Так, например, С-реактивный белок может связать до 90% циркулирующих апоВ-ЛП [32,53].
Острофазная реакция печени и циркуляция острофазных белков в свою очередь ведут к активации и последующей дисрегуляции системы мононуклерных фагоцитов и системы лимфоидной ткани организма. Циркулирующие комплексы С-реактивный белок + ЛП, избыток ХС и ЛПНП и особенно м-ЛПНП, в условиях экспрессии молекул адгезии сосудистого эндотелия (VCAM), повреждают интиму сосуда, откладываются в ней (преимущественно в виде м-ЛПНП) [31,52] и становясь аутоантигенами, запускают местную - воспалительную и системную аутоиммунную реакции [31,33].
Активированные макрофаги, мигрирующие в сосудистую стенку, презентируют аутоантиген, активируют эндотелиоциты и начинают секретировать цитокины, ростовые факторы, кинины и другие медиаторы, привлекая в образующийся очаг воспаления клетки иммунной системы. Постоянная выработка цитокинов и факторов роста, на фоне продолжающегося отложения м-ЛПНП и ХС, приводят к пролиферации и миграции гладкомышечных клеток медии, повышенной секреции коллагена и развитию фиброза сосудистой стенки [31,33,43,44]. В конечном итоге формируется типичная атероматозная бляшка, которая относится к поздним атеросклеротическим изменениям.
При атеросклерозе обнаруживаются и системные иммунные сдвиги, обусловленные дисбалансом активности клеток лимфоидной ткани. Наблюдается снижение активности Т-супрессорного звена и активация В-звена иммунитета. In vitro отмечается двукратное снижение пролиферативного ответа лимфоцитов крови на митоген. То же самое наблюдается и при действии на них ЛПОНП больных атеросклерозом людей с ДЛП [8]. Однако, в Т-зависимых зонах селезёнки и лимфоузлов происходит активация лимфоцитогенеза (возрастает митотическая активность, увеличивается количество лимфобластов) [31], и это может указывать на блок оттока бластных клеток из органов иммуногенеза, т.е. на дисрегуляцию иммунных процессов в организме.
Всё вышеизложенное даёт право заключить, что атеросклероз – это хроническая системная воспалительная реакция организма со специфическими локальными проявлениями на стенке сосудов. Локальные проявления атеросклероза развиваются на фоне ДЛП (выражающейся нарушением соотношения отдельных фракций ЛП и липидов) в условиях дисфункции печени и иммунной системы и сопровождаются необязательным повышением концентраций ЛП и ХС в крови [25,31].
АТЕРОСКЛЕРОЗ
Атеросклероз – хроническое прогрессирующее заболевание крупных и средних эластических и мышечно-эластических артерий. Атеросклероз характеризуется пролиферативно-синтетическим ответом ряда клеток сосудистой стенки и крови – гладкомышечных макрофагов, тромбоцитов, фибробластов на патологические (качественно своеобразные или количественно избыточные) ЛП, с формированием в интиме фиброатером.
Причины развития атеросклероза:
1. Гиперхолестеринемия;
2. Гиперлипидемия ЛПОНП, ЛППП и ЛПНП (вызывают генетические дефекты рецепторов, апобелков, СД, гипотериоз, переедание).
3. Изменение нормальной структуры ЛПНП под действием ПОЛ и гипергликемии. Избыток глюкозы гликозилирует апобелки, повышенное ПОЛ (при гипоксии, воспалении) повреждает липиды и апобелки ЛП. Модифицированные ЛПНП становятся чужеродными для организма, атакуются антителами и поглощаются макрофагами с участием «скевенджер-рецепторов» (рецепторов-мусорщиков);
4. Повреждение сосудистой стенки высоким артериальным давлением (психоэмоциональные стрессы), ПОЛ (гипоксия, курение (через СО), воспаления), иммунными реакциями, токсинами и другими ядовитыми веществами (Pb, Cd). Повреждающие факторы разрыхляют и истончают (до исчезновения) гликокаликс энтероцитов, увеличивают межэндотелиальные щели, что создает на поверхности эндотелия зоны повышенной клейкости и проницаемости;
5. Принадлежность к мужскому полу (гормональный статус).
Молекулярные механизмы развития атеросклероза
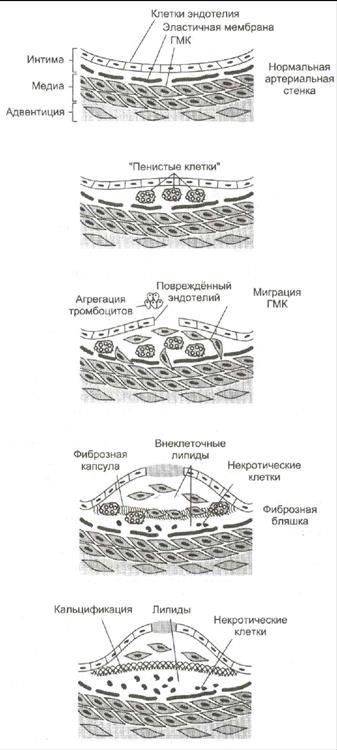
Развитие атеросклероза проходит в 6 стадий:
1. Стадия измененного эндотелия. На поверхности поврежденного эндотелия скапливаются тромбоциты и моноциты. Модифицированные ЛПНП проникают под поврежденный эндотелий сосудов. За ними направляются моноциты (в ткани они макрофаги) и захватывают ЛП через скевенджер-рецепторы. Этот процесс не ингибируется избытком ХС, поэтому макрофаги перегружаются ХС и превращаются в «пенистые клетки». Отдельные «пенистые клетки» есть у новорожденных.
2. Стадия жировых полосок. При увеличении количества «пенистых клеток» они образуют липидные полоски. «Пенистые» клетки адсорбируют все остальные липиды без разбора. Поврежденный эндотелий, активированные макрофаги, тромбоциты выделяют БАВ, которые стимулируют пролиферацию ГМК и миграцию их в очаг повреждения.
3. Стадия переходная. Активированные ГМК синтезируют коллаген и эластин, что приводит к прорастанию бляшки фиброзной тканью. Клетки под фиброзной оболочкой некротизируются, а ХС начинает откладываться в межклеточном пространстве. Может происходить разрыв эндотелия сосудов.
4. Стадия атеромы. ХС межклеточного пространства формирует в центре бляшки липидную каплю – атерому, которая через разрушенный эндотелий выступает в просвет сосуда.
5. Стадия фиброатеромы. Атерома пропитываясь солями кальция, белками, ГАГ и приобретает плотную фиброзную крышку. Атерома становиться фиброатеромой.
6. Стадия осложнения фиброатеромы. Фиброатерома не стабильна, она может надрываться и изъявляться, что приводит к обострению атеросклероза.
Осложнения. Поврежденный эндотелий прекращает синтез PGI2, который в норме ингибирует тромбоциты. Тромбоциты активируются и секретируют тромбоксан ТХА2 и тромбоцитарный фактор роста (пептид). Тромбоцитарный фактор роста привлекает в бляшку клетки крови, ГМК, что способствует росту бляшки и развитию очага воспаления. ТХА2 → агрегацию тромбоцитов → образование тромбов → закупорка сосудов → ишемия тканей → некроз тканей → изъявления стенок сосудов → кровотечения, аневризмы. Оторвавшиеся тромбы → эмболии сосудов.
Чаще всего атеросклероз развивается в коронарных, мозговых, почечных артериях, артериях нижних конечностей и в аорте. Атеросклероз коронарных артерий проявляется ИБС, мозговых – ИБ мозга, почек – вазоренальной артериальной гипертензией. Спазм или тромбоз коронарных сосудов ведет к инфаркту миокарда, эмболия сонных артерий ведет к развитию инсультов.
Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения.
Биохимические основы лечения атеросклероза
Лечение гиперхолестеролемии, как правило, комплексное.
I Диета. Необходимо употреблять:
1) продукты гипокалорийные, гипохолестериные, с низким содержанием легкоусвояемых углеводов (растительная пища). Поступление ХС с пищей не должно превышать 0,3 мг/сут;
2) полиеновые ЖК семейства ω-3 (морепродукты). Из них синтезируются простагландины, подавляющие тромбообразование и замедляют развитие атеросклеротической бляшки. Ненасыщенные ЖК также ускоряют выведение ХС из организма (механизм не ясен);
3) витамины С, Е, А и другие антиоксиданты ингибирующие ПОЛ и поддерживающие нормальную структуру ЛПНП и их метаболизм.
Липримал дает самый сильный эффект
II. «Размыкание» цикла энтерогепатической циркуляции жёлчных кислот. Лекарства типа холестирамина, холестипол (полимеры) адсорбируют в кишечнике жёлчные кислоты, выделяются с фекалиями и таким образом уменьшают возврат жёлчных кислот в печень. В печени увеличивается захват ХС из крови для синтеза новых жёлчных кислот.
III. Ингибирование синтеза ХС. Наиболее эффективные препараты для лечения атеросклероза — ингибиторы ГМГ-КоА-редуктазы, например антибиотик мевакор. Такие препараты могут почти полностью подавить синтез ХС в организме, нормализуя уровень ХС.
IV. Активация катаболизма ЛП. Лекарственные препараты — фибраты (клофибрат, фенофибрат) активируют ЛПЛ и ускоряют катаболизм ЛПОНП. Эти препараты также активируют окисление ЖК в печени, уменьшая тем самым синтез ТГ и ЭХС и, как следствие, секрецию ЛПОНП печенью.
Для эффективного лечения атеросклероза применяют, как правило, комбинированное воздействие нескольких лекарственных препаратов.
| |
Берсенёв Алексей Вячеславович диссертация
По современным представлениям атеросклероз – это хроническая системная воспалительная реакция организма, развивающаяся на фоне дислипидемии и сопровождающаяся образованием одиночных или множественных очагов липидных отложений (атероматозных бляшек) на внутренней поверхности сосудов [134].
Полагают, что именно системная воспалительная реакция способствует развитию дислипидемии (ДЛП) и запускает процесс атерогенеза [33,53]. В свою очередь алиментарные и наследственные ДЛП также индуцируют проявления синдрома системного воспалительного ответа и усугубляют тяжесть атеросклеротического поражения сосудов в организме [12,13,18,22,43].
В присутствии провоспалительных факторов, таких как окисленные ЛП (в особенности низкой и очень низкой плотности) [16,18,43], инфекционные агенты [33,53] и различные неспецифические стресс-факторы, в организме активируется макрофагально-моноцитарная система и усиливается выработка провоспалительных цитокинов (интерлейкинов: IL-1, 6, фактора некроза опухоли: TNF-α и др.). Эти цитокины, с одной стороны, вызывают в сосудистом эндотелии экспрессию молекул адгезии – ICAM-1, ICAM-2 (intracellular adhesion molecules), VCAM-1 (vascular cell adhesion molecules), селектины и др. и нарушают структуру эндотелиальной выстилки сосудов, а с другой вызывают экспрессию в гепатоцитах генов, ответственных за синтез в печени острофазных белков. Участие печени в острофазном процессе и, следовательно, в инициации и модуляции системной воспалительной реакции организма [66,134], смещает в ней баланс биохимических механизмов, что вызывает прежде всего, нарушения липидного обмена, поскольку печень играет центральную роль в регуляции этого вида обмена в организме.
Длительно поддерживаемая в организме активация макрофагально-моноцитарной системы из адаптивной постепенно превращается в повреждающую, при которой не только нарушается регуляция печенью липидного обмена, но и создаются условия для прогрессирования ДЛП и атеросклероза, а также для развития их осложнений [12,13,22].
При ДЛП и атеросклерозе клетками-мишенями (при системной воспалительной реакции) являются, прежде всего, клетки печени – гепатоциты, купферовские клетки, эндотелиоциты, а также эндотелиальная выстилка сосудов [16,17], изменения в которых развиваются параллельно, постепенно прогрессируют, ведут к формированию хронического гепатита [17], а также к типичному повреждению сосудистой стенки атеросклеротическим процессом.
ЖЕЛЧЕКАМЕННАЯ БОЛЕЗНЬ
Желчнокаменная болезнь — патологический процесс, при котором в жёлчном пузыре образуются камни, основу которых составляет ХС.
Выделение ХС в жёлчь должно сопровождаться пропорциональным выделением жёлчных кислот и фосфолипидов, удерживающих гидрофобные молекулы ХС в жёлчи в мицеллярном состоянии.
Если активность ГМГ-КоА-редуктазы повышена, а активность 7-а-гидроксилазы снижена - ХС синтезируется много, а жёлчных кислот мало. Это приводит к диспропорции ХС и жёлчных кислот, секретируемых в жёлчь. ХС начинает осаждаться в жёлчном пузыре, образуя вначале вязкий осадок, который постепенно становится более твёрдым. Иногда он пропитывается билирубином, белками и солями кальция. Камни, образующиеся в жёлчном пузыре, могут состоять только из ХС (холестериновые камни) или из смеси ХС, билирубина, белков и кальция.
Холестериновые камни обычно белого цвета, а смешанные камни — коричневого цвета разных оттенков.
Причин, приводящих к изменению соотношения жёлчных кислот и ХС, в жёлчи много: пища, богатая ХС, гиперкалорийное питание, застой жёлчи в жёлчном пузыре, нарушение энтерогепатической циркуляции, нарушения синтеза жёлчных кислот, инфекции жёлчного пузыря.
Если камни начинают перемещаться из жёлчного пузыря в жёлчные протоки, то они вызывают спазм жёлчного пузыря и протоков, что больной ощущает как приступ сильной боли. Если камень перекрывает проток некоторое время, то нарушается поступление жёлчи в кишечник, жёлчные пигменты проходят через мембраны гепатоцитов в сторону синусоидов и попадают в кровь, что приводит к развитию обтурационной (подпечёночной желтухи).
Лечение желчнокаменной болезни
В начальной стадии образования камней можно применять в качестве лекарства хенодезоксихолевую кислоту. Попадая в жёлчный пузырь, эта жёлчная кислота постепенно растворяет осадок ХС (холестериновые камни), однако это медленный процесс, требующий нескольких месяцев.
Список литературы
Берсенёв Алексей Вячеславович. Кандидатская диссертация: Трансплантация клеток эмбриональной печени и стволовых клеток костного мозга для коррекции дислипидемии и ранних стадий атерогенеза. М.: 2003.
Дата добавления: 2015-07-18; просмотров: 1122;