КЕТОГЕНЕЗ И УТИЛИЗАЦИЯ КЕТОНОВЫХ ТЕЛ
При повышенном окислении жирных кислот в печени скорость, с которой образуется ацетил-СоА, может превышать скорость его окисления в цикле ТКК. В таких условиях ацетил-СоА используется для продукции ацетоуксусной, b-оксимасляной кислот и ацетона. Эти вещества носят собирательное название кетоновых тел. Присутствуя в избыточных количествах, например при голодании или некомпенсированном диабете, ацетоацетат и b-оксибутират обусловливают метаболический ацидоз (т. е. «голодательный» кетоз или диабетический кетоацидоз). Большие количества ацетона, имеющиеся при различных формах кетоза, не играют роли в развитии метаболического ацидоза, но способствуют появлению у больного характерного фруктового запаха при дыхании.
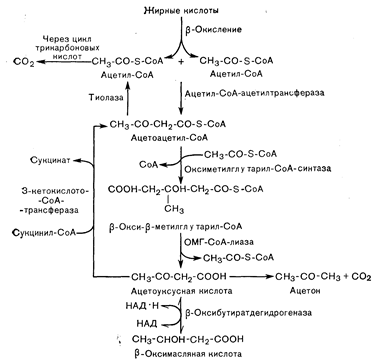
Рис.10—7. Метаболические пути образования кетоновых тел (ацетоуксусной и b-оксимасляной кислоты) из ацетил-СоА и их окисления до СО2. Ограничивающая скорость стадия образования кетонов предшествует образованию ацетил-СоА и локализуется на этапе переноса жирного ацил-СоА через митохондриальную мембрану к месту расположения ферментов, участвующих в b-окислении (см. рис. 10—8). Образование ацетоуксусной кислоты из ацетоацетил-СоА требует вначале образования b-окси-бета-метилглутарил-СоА.
Синтез кетоновых тел из ацетил-СоА (рис. 10—7) в качестве начального этапа предполагает конденсацию двух молекул ацетил-СоА с образованием ацетоацетил-СоА. Превращение ацетоацетил-СоА в ацетоуксусную кислоту требует его начального превращения в b-окси-бета-метилглютарил-СоА, который расщепляется на ацетоуксусную кислоту и ацетил-СоА. Образование b-оксибутирата из ацетоацетата включает обратимую окислительно-восстановительную реакцию, кофактором которой служит НАД•Н. Ацетон образуется из ацетоацетата путем неферментативного спонтанного декарбоксилирования.
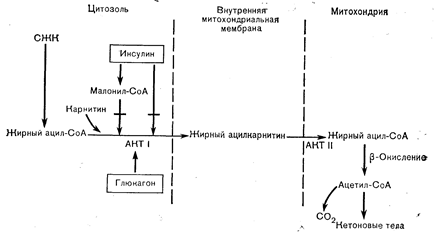
Рис. 10—8. Внутрипеченочная регуляция кетогенеза. Главным ограничивающим скорость этапом является перенос жирных ацильных производных через митохондриальную мембрану, осуществляемый ферментами ацилкарнитинтрансферазой (АКТ) I и II. АКТ I тормозится при повышения уровня инсулина, уменьшающего доступность карнитинаа и увеличивающего .доступность малонил-СоА—прямого ингибитора АКТ I. Наоборот, снижение уровня инсулина и повышение уровня глюкагона приводит к увеличению содержания карнитина, снижению содержания малонил-СоА и повышению активности АКТ-I (по McGarry J. D., Diabetes, 1979, 28, 517, с модификациями). СЖК — свободные жирные кислоты.
Что касается факторов, регулирующих скорость кетогенеза, то важнейшим из них, очевидно, является увеличение доставки жирных кислот в печень (повышенный липолиз). Однако доставка субстрата — это не единственная детерминанта скорости процесса, поскольку у здоровых лиц повышение уровня СЖК в крови, вызываемое приемом жирной (содержащей триглицериды) пищи одновременно с введением активатора липопротеиновой липазы (гепарин), не приводит к кетозу (17а). Вторым требованием является увеличение скорости b-окисления в митохондриях печени. Только при этом поступающие в печень жирные кислоты будут использоваться для образования ацетил-СоА, а не для синтеза триглицеридов. Как уже отмечалось, ограничивающей скорость стадией b-окисления жирных кислот является ацилкарнитинтрансферазная реакция. Эта реакция ускоряется при повышении уровня свободного карнитина и снижении уровня малонил-СоА — ближайшего интермедиата в процессе синтеза жира. Оба эти условия (повышение уровня карнитина в печени и уменьшение содержания малонил-СоА) выполняются при снижении уровня инсулина и в некоторой степени при увеличении содержания глюкагона [15]. Таким образом, кетогенез стимулируется как повышенным липолизом в жировой ткани, так и активацией процесса b-окисления в печени (рис. 10—8).
При повышенной продукции кетоновых тел в печени не обязательно тормозится эстерификация жирных кислот (т. е. образование триглицеридов) или окисление ацетил-СоА в цикле ТКК. Больше того, на фоне повышенной продукции печенью кетоновых тел образование триглицеридов может даже увеличиться, если резко увеличивается доставка жирных кислот. Вследствие этого при недостаточном компенсированном диабете частой находкой является ожиревшая, перегруженная триглицеридами печень. С другой стороны, при повышенной продукции кетоновых тел в печени синтез жирных кислот из ацетил-СоА не происходит.
Образуемые в печени ацетоацетат и b-оксибутират попадают в кровоток и циркулируют в отношении примерно 1:3 соответственно. Циркулирующие кетоновые тела подвергаются окислению в мышечной ткани, а в условиях очень длительного голодания поглощаются и окисляются в ткани мозга [16]. Первый этап распада кетоновых тел — это окисление b-оксибутирата в ацетоацетат. Затем в процессе трансферазной реакции с сукцинил-СоА или с помощью тиокиназы, требующей АТФ, образуется ацетоацетил-СоА. Последний распадается с образованием двух молекул ацетил-СоА (см. рис. 10—7). Эти реакции происходят в мышце, головном мозге и зародышевой, но не зрелой печени.
Дата добавления: 2015-01-19; просмотров: 3795;