Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕН
18 Эта* 552
| Рис. 81. Структура гликогена |
Клетки нервной системы зависят от глюкозы как от основного энергетического субстрата. В то же время в мозге нет запасов глюкозы, она там не синтезируется, нейроны не могут потреблять другие энергетические субстраты кроме глюкозы и кетоновых тел.
Гликоген. Из Г-6-Ф в результате сочетанного действия гликогенсинтетазы и «ветвящего» фермента синтезируется гликоген - полимер, напоминающий по виду дерево. В молекуле гликогена может содержаться до миллиона моносахаридов. При этом происходит как бы кристаллизация гликогена и он не обладает осмотическим эффектом. Такая форма пригодна для хранения в клетке. Если бы такое количество молекул глюкозы было растворено, то из-за осмотических сил клетку бы разорвало. Гликоген является депонированной формой глюкозы. Он содержится практически во всех тканях; в клетках нервной системы его количество минимально, а в печени и мышцах его особенно много. Гликоген содержит только два типа гликозидных связей: а(1-»4)-тип и а(1-»6)-тип. Связь а(1->4)-тип формируется через каждые 8-10 остатков D-глю-козы (рис. 81).
Гликогенолиз. Это путь расщепления гликогена. Гликоген в организме в основном сохраняется в печени и скелетных мышцах. Гликоген мышц используется в качестве источника энергии при интенсивной физической нагрузке. Гликогенолиз в печени активируется в ответ на снижение концентрации глюкозы при перерывах в приеме пищи или в ответ на стрессовые воздействия. Основными гормонами, активирующими гликогенолиз, являются глюкагон, адреналин (эпинефрин) и кЪртизол (табл. 36).
Снижение синтеза гликогена. Снижение синтеза происходит при поражении гепатоцитов (ге-
Гормональная регуляция гликогенолиза
Таблица 36
| Гормон | Место образования | Инициатор | Эффект на гликогенолиз |
| Глюкагон | а-клетки поджелудочной железы | Гипогликемия | Быстрая активация |
| Адреналин | Мозговой слой | Стресс, | Быстрая активация |
| надпочечника | гипогликемия | ||
| Кортизол | Кора надпочечников | Стресс | Длительная активация |
| Инсулин | р-клетки поджелудочной железы | Гипергликемия | Подавление |
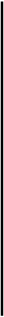 патиты, отравление фосфором, четыреххлорис-тым углеродом и др.); гипоксии, когда дефицит кислорода неизбежно приводит к существенному снижению эффективности образования АТФ, необходимого для синтетических процессов; снижении тонуса парасимпатической нервной системы; гиповитаминозах В: и С; эндокринных заболеваниях - сахарном диабете, тиреотоксикозе, недостаточности надпочечников (болезнь Ад-дисона).
патиты, отравление фосфором, четыреххлорис-тым углеродом и др.); гипоксии, когда дефицит кислорода неизбежно приводит к существенному снижению эффективности образования АТФ, необходимого для синтетических процессов; снижении тонуса парасимпатической нервной системы; гиповитаминозах В: и С; эндокринных заболеваниях - сахарном диабете, тиреотоксикозе, недостаточности надпочечников (болезнь Ад-дисона).
Усиление распада гликогена.Усиление гликогенолиза в печени происходит при возбуждении центральной нервной системы. Нервные импульсы проводятся симпатической нервной
| "Вертящий" фермент |
 Синтез гликогена
Синтез гликогена
Рис. 82. Дефекты ферментов метаболизма гликогена, приводящие к гликогенозам разного типа
системой к депо гликогена и активируют процесс его распада, обеспечивая поступление глюкозы в кровь. Усиление гликогенолиза наблюдается также при повышении продукции гормонов - стимуляторов гликогенолиза (адреналина, глюкагона, тироксина и соматотропного гормона) и при интенсивной мышечной работе, что обусловливается увеличением потребления глюкозы мышцами. Кроме того, распад гликогена повышается при шоке, лихорадке, эмоциональных нагрузках.
При недостаточности гликогена (вследствие снижения синтеза или уменьшения его распада) тканевая энергетика переходит на использоване в качестве субстратов для окисления жиров и белков. В результате этого происходит избыточное образование кетоновых тел иразвивается интоксикация. Использование клеткой белков как источника энергии обусловливает нарушения различных ферментативных и пластических процессов.
Гликогенозы.Так называются болезни патологического депонирования гликогена. Это группа наследственных патологий, при которых вследствие генетически обусловленных дефектов некоторых ферментов метаболизма гликогена происходит его избыточное накопление в различных органах, прежде всего в печени и скелетных мышцах. При некоторых типах гликогено-зов синтезируется гликоген с нарушенной структурой (рис. 82).
В настоящее время известны 12 типов гли-когенозов. Наиболее часто встречаются следующие:
1. Гликогеноз I типа, или болезнь Гирке (Крефельда - Гирке), или гепатонефромегаль-ный.Впервые описан в 1929 г. Этот тип глико-геноза встречается наиболее часто. В основе данной патологии лежит наследственный дефицит фермента глюкозо-6-фосфатазы в печени и поч-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
ках. Под действием этого фермента происходит дефосфорилирование глюкозо-6-фосфата, что обеспечивает трансмембранный переход свободной глюкозы из гепатоцитов и клеток почек в кровь (рис. 82). Дефицит фермента вызывает накопление избытка гликогена в клетках печени и почек. Структура гликогена при этом не нарушается. У больных увеличен живот за счет существенного увеличения размеров печени (гепа-томегалия). Содержание гликогена в печени достигает 10-15% от массы органа (вместо 3-5% в норме). В связи с дефектом фермента поступление в кровь глюкозы значительно сокращается, что обусловливает тяжелую гипогликемию, являющуюся причиной приступов судорог. Дефицит глюкозы в крови приводит к торможению выделения инсулина из поджелудочной железы, что стимулирует липолиз в жировой ткани. Формируется характерная для данного типа гли-когеноза гиперлипидемия. Интенсификация метаболизма липидов приводит к накоплению в крови молочной кислоты и гидроксибутирата (кетоз). Дефицит инсулина также приводит к снижению интенсивности синтеза белка (нарушается транспорт аминокислот). Пациенты, страдающие этим типом гликогеноза, как правило, рано умирают от интеркуррентных заболеваний или от ацидотической комы.
2. Гликогенов II типа, или болезнь Помпе. Развивается при наследственном дефекте фермента а-1,4-глюкозидазы, локализованного в ли-зосомах. Фермент катализирует расщепление гликогена, а освобождаемая глюкоза выходит из лизосом в цитоплазму и становится составной частью пула глюкозы клетки. Дефект фермента приводит к генерализованному накоплению гликогена с нормальной структурой в печени, селезенке, почках, скелетных и сердечной мышцах, нервной ткани, лейкоцитах и эритроцитах. Клинические симптомы заболевания проявляются уже на 2-6-м мес жизни. Наиболее тяжелые нарушения развиваются в мышечной ткани: гипотония скелетных мышц и увеличение размеров сердца (кардиомегалия), что ведет к тяжелой кардиореспираторной недостаточности. Она является главной причиной летального исхода в зозрасте до 2 лет.
3. Гликогеноз III типа, или болезнь Кори (Кори и Форбса). Заболевание возникает по причине наследственного дефекта фермента амило-1,6-глюкозидазы мышц, печени и миокарда (рис.
82) Молекула полимера при данном типе гликогеноза имеет патологическую структуру, для которой характерны многочисленные, но укороченные боковые ветви. Такой гликоген в избытке накапливается преимущественно в печени, мышечной ткани, эритроцитах и зернистых лейкоцитах. Затрудненный гидролиз гликогена с измененной структурой приводит к гипогликемии. Имеет место гиперлипидемия, развивающаяся по механизму, аналогичному для гликогеноза I типа, но в более мягкой форме. Угрозы для жизни не представляет. Специфическим диагностическим признаком гликогеноза III типа является повышенный уровень гликогена в зернистых лейкоцитах.
4. Гликогеноз IV типа, или болезнь Андерсена. В основе заболевания лежит наследственный дефицит фермента a-D-1,4 глюкан 6-а-глю-козилтрансферазы, который обеспечивает ветвление в молекуле гликогена. Недостаточность фермента обусловливает накопление преимущественно в печени, мышцах и лейкоцитах гликогена, молекулы которого имеют аномально длинные цепи с очень малым количеством боковых ответвлений, делающим ее структуру похожей на таковую у крахмала. Для клинической картины данного заболевания характерны: выраженная гипогликемия, гепатомегалия, прогрессирующий цирроз печени с летальным исходом вследствие печеночной недостаточности в возрасте до 2 лет.
Помимо приведенных выше типов, описаны более редкие, а также смешанные гликогенозы: V тип, или болезнь Мак-Ардля (Мак-Ардля -Шмида - Пирсона); VI тип, или болезнь Герса; VII тип, или болезнь Таруи; VIII тип, или болезнь Ходжина, и другие.
11.4.3. Нарушения промежуточного обмена углеводов
Причинами нарушения промежуточного Обмена углеводов могут быть:
1. Гипоксия. Вызывается недостаточностью кровообращения, дыхания и др. Развивающийся дефицит кислорода переключает клеточный метаболизм с аэробного на анаэробный тип, при котором основным источником энергии становится анаэробный гликолиз. При распаде глюкозы в этих условиях образуется избыток молочной и пировиноградной кислот. Молочная кислота в
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
тканях способствует усилению диссоциации ок-сигемоглобина и расширению коронарных сосудов, компенсируя, таким образом, явления гипоксии. В норме молочная кислота из тканей, где идет гликолиз (например, из мышц), поступает с кровью в печень (цикл Кори), где превращается при участии фермента лактатдегидроге-назы в пируват. Пируват в печени частично окисляется, а частично превращается в глюкозу (глю-конеогенез). Таким образом лактат возвращается в метаболический фонд углеводов. Длительное существование избытка молочной кислоты в тканях приводит к дефициту субстрата окисления - глюкозы, что вызывает дальнейшее снижение эффективности синтеза АТФ. Дефицит макроэргов лежит в основе нарушения трансмембранного переноса ионов и повышения проницаемости мембран. В конечном итоге это приводит к значительным структурно-функциональным повреждениям в тканях, вплоть до гибели клеток.
2. Нарушения функций печени. В гепатоци-тах часть молочной кислоты в норме ресинтези-руется в глюкозу и гликоген. При поражении печени этот процесс нарушается, молочная кислота выходит в кровь, развивается ацидоз.
3. Гиповитаминоз В,. Витамин В; (тиамин) в результате процесса фосфорилирования превращается в кокарбоксилазу - простетическую группу ряда ферментов углеводного обмена. При недостаточности витамина В, возникает дефицит кокарбоксилазы, что приводит к подавлению синтеза ацетил-КоА из пировиноградной кислоты. Последняя накапливается и частично переходит в молочную кислоту, содержание которой в связи с этим возрастает. Торможение окисления пировиноградной кислоты снижает синтез ацетилхолина, что вызывает нарушение передачи нервных импульсов. При возрастании концентрации пировиноградной кислоты в 2-3 раза по сравнению с нормой возникают нарушения чувствительности, невриты, параличи и др. Гиповитаминоз Bt приводит также к нарушению работы пентозофосфатного пути окисления вследствие понижения активности фермента транске-толазы.
11.4.4. Гипергликемические состояния
Уровень глюкозы в крови является важнейшим фактором гомеостаза. Он поддерживается
на определенном уровне функцией кишечника, печени, почек, поджелудочной железы, надпочечников, жировой ткани и других органов (рис. 83).
Выделяют несколько типов регуляции углеводного обмена: субстратную, нервную, почечную, гормональную.
Субстратная регуляция. Основным фактором, определяющим метаболизм глюкозы, является уровень гликемии. Пограничная концентрация глюкозы, при которой продукция ее в печени равна потреблению периферическими тканями, составляет 5,5-5,8 ммоль/л. При уровне, меньшем этого, печень поставляет глюкозу в кровь; при большем уровне, наоборот, доминирует синтез гликогена в печени и мышцах.
Нервная регуляция. Возбуждение симпатических нервных волокон приводит к освобождению адреналина из надпочечников, который стимулирует расщепление гликогена в процессе гли-когенолиза. Поэтому при раздражении симпатической нервной системы наблюдается гипер-гликемический эффект. Наоборот, раздражение парасимпатических нервных волокон сопровождается усилением выделения инсулина поджелудочной железой, поступлением глюкозы в клетку и гипогликемическим эффектом.
Почечная регуляция.В клубочках почек глюкоза фильтруется, затем в проксимальных канальцах реабсорбируется энергозависимым механизмом. Величина канальцевой реабсорбции относительно постоянна, с возрастом имеется тенденция к снижению. При превышении в сыворотке уровня 8,8 - 9,9 ммоль/л глюкоза выделяется с мочой. Показатель гликемии, при котором появляется глюкозурия, называется почечным порогом. На выделение глюкозы с мочой влияет скорость клубочковой фильтрации, которая в норме составляет примерно 130 мл/мин. При снижении фильтрации при почечной недостаточности или уменьшении кровоснабжения почек глюкоза будет отсутствовать в моче даже при гликемии, значительно превышающей почечный порог, так как фильтруется меньше глюкозы и вся она успевает реабсорбироваться в проксимальных канальцах почек. В случае не-фропатий с нарушением реабсорбции глюкоза может появиться в моче даже при нормоглике-мии. Поэтому по уровню глюкозы в моче нельзя ставить диагноз сахарный диабет.
Гормональная регуляция.На уровень глюкозы в крови влияет широкий спектр гормонов,
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
при этом практически только инсулин вызывает гипогликемический эффект. Контринсуляр-ным действием с повышением уровня глюкозы крови обладают глюкагон, адреналин, глюкокор-тикоиды, СТГ, АКТГ, ТТГ. Эффекты инсулина и контринсулярных гормонов в норме контролируют достаточно стабильный уровень глюкозы в крови. При низкой концентрации инсулина, в частности при голодании, усиливаются гипер-гликемические эффекты других гормонов, таких как гормон роста, глюкокортикоиды, адреналин и глюкагон. Это происходит даже в том случае, если концентрация этих гормонов в системе циркуляции не увеличивается. В табл. 37 представлены основные эффекты гормонов на метаболизм глюкозы.
Физиологически в регуляции обмена глюкозы наиболее важны два гормона - инсулин и глюкагон.
Инсулин - полипептид, состоит из двух цепей: А-цепь содержит 21 аминокислоту, В-цепь - 30 аминокислот. Цепи соединены между собой 2 дисульфидными мостиками. Инсулин схож у разных видов млекопитающих: так, А-цепь иден-
тична у человека, свиньи, собаки, кашалота; В-цепь идентична у быка, свиньи и козы. Фактически инсулин человека и свиньи отличаются только тем, что на карбоксильном конце В-цепи у свиньи находится аминокислота аланин, а у человека треонин. Поэтому коммерческий «человеческий инсулин» производится путем замены аланина на треонин в инсулине свиньи.
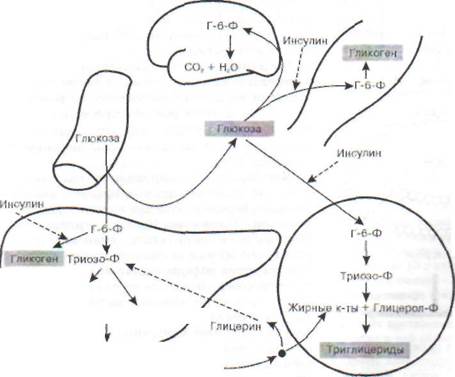
Инсулин синтезируется в виде неактивной полипептидной цепи проинсулина, таким он сохраняется в гранулах (3-клеток островков Лан-герганса поджелудочной железы. Активация проинсулина заключается в частичном протео-лизе пептида по Arg31 и Arg63 (рис. 84). В результате в эквимолярном количестве образуются инсулин и С-пептид (connecting peptide).
Инсулин в крови находится в свободном и связанном с белками состоянии. Деградация инсулина происходит в печени (до 80%), почках и жировой ткани. С-пептид также подвергается деградации в печени, но значительно медленнее. Базальная концентрация инсулина, определяемая радиоиммунологически, составляет у здоровых 15-20 мкЕд/мл. После пероральной
| Мышцы |
| Мозг |
| Кишечник |
 АцетилКоА
АцетилКоА
Жирные к-ты + Глицерол-Ф
Триглицериды
Печень
ЛПОНГ1
Жировая ткань
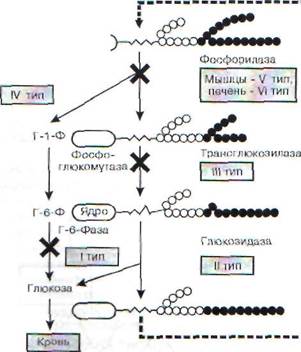
Рис. 83. Метаболизм глюкозы после еды. Всосавшаяся в кишечнике глюкоза поступает в печень. Печень поддерживает постоянную доставку энергетических субстратов для других
органов, в частности мозга. Поступление глюкозы в печень и мозг не зависит от инсулина, в мышцы и жировую ткань -инсулинзависимое. Во всех клетках первый этап метаболизма глюкозы - образование глюкозо-6-фосфата. В печени инсулин стимулирует фермент глюкокиназу, переводя Г-6-Ф в гликоген, избыток Г-6-Ф переводится в жирные кислоты с последующим образованием триглицеридов, которые освобождаются из печени в виде липопротеидов очень низкой плотности (ЛПОНП). В мышцах глюкоза запасается в виде гликогена, в жировой ткани переходит в триглицериды, в мозговой ткани глюкоза используется как энергетический субстрат
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
Гормоны, контролирующие гомеостаз глюкозы
Таблица 37
| Гормон | Механизм действия | Ткань |
| Инсулин | Увеличивает: потребление глюкозы клетками | Мышцы, жировая ткань |
| синтез гликогена | Печень, мышцы | |
| синтез белков | Печень, мышцы | |
| синтез жирных к-т и триглицеридов | Печень, жировая ткань | |
| снижает: глюконеогенез | Печень | |
| кетогенез | Печень | |
| липолиз | Жировая ткань | |
| протеолиз | Мышцы | |
| Глюкагон | Увеличивает: гликогенолиз | Печень |
| глюконеогенез | Печень | |
| кетогенез | Печень | |
| липолиз | Жировая ткань | |
| Адреналин | Увеличивает: гликогенолиз | Печень, мышцы |
| липолиз | Жировая ткань | |
| Гормон роста | Увеличивает: гликогенолиз | Печень |
| липолиз | Жировая ткань | |
| Кортизол | Увеличивает: глюконеогенез | Печень |
| синтез гликогена | Печень | |
| протеолиз | Мышцы | |
| Снижает: потребление глюкозы клетками | Мышцы, жировая ткань |
нагрузки глюкозой уровень его через 1 ч повышается в 5-10 раз по сравнению с исходным. Скорость секреции инсулина натощак составляет 0,5-1 Ед/ч, после приема пищи увеличивается до 2,5-5 Ед/ч. У здоровых людей наблюдают-
Соединяющий пептид (С-пептид)
Рис. 84. Образование инсулина в поджелудочной
железе. В результате частичного протеолиза проин-
сулина формируются инсулин и С-пептид. Инсулин
состоит из двух полипептидных цепей, соединенных
дисульфидными мостиками
ся две фазы секреции инсулина - ранний пик (через 3-10 мин после углеводной нагрузки) и поздний пик (через 20 мин). Раннее выделение инсулина сдерживает резкий подъем глюкозы при ее всасывании.
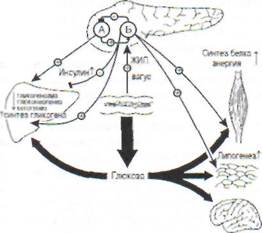
Секреция инсулина стимулируется, помимо гипергликемии, глюкагоном, а также полипептидными гормонами кишечника, включая желудочно-кишечный инсулинотропный полипептидный гормон (ЖИП), аминокислотами, свободными жирными кислотами, раздражением ва-гуса (рис. 85).
Метаболическое действие инсулина комплексное, оно включает прямые эффекты на обмен липидов, белков и особенно в связи с сахарным диабетом - D-глюкозы. Инсулин усиливает мембранный транспорт глюкозы, аминокислот и К+, активирует многие внутриклеточные ферменты. В то же время полипептидная молекула инсулина не способна проникать через клеточную мембрану, поэтому все эффекты инсулина осуществляются через специальные рецепторы на поверхности клеточной мембраны. Инсулиновый рецептор комплексный, он состоит из а- и 0-субъ-единиц, соединенных дисульфидными мостиками.
Высокие концентрации инсулина в крови обладают анаболическим, а низкие - катаболичес-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
|
| тглюкагсн |
Рис. 85. Гомеостаз глюкозы у здорового человека.
Во время приема пищи секреция инсулина Р-клетками поджелудочной железы увеличивается подвлиянием желудочно-кишечного инсулинотроп-
ного полипептидного гормона (ЖИП) и вагусных
стимулов. Инсулин подавляет секрецию глюкагона
поджелудочной железой и синтез глюкозы печенью.
Одновременно инсулин стимулирует поглощение
глюкозы в инсулинзависимых органах (печень,
скелетные мышцы, жировая ткань). В инсулиноне-
зависимые органы (мозг, периферические нервы,
эритроциты, кровеносные сосуды, соединительная
ткань, почки) поступление глюкозы зависит от
уровня ее в системе кровотока. При повышении
отношения инсулин/глюкагон (после еды) глюкоза
запасается в гликогене и превращается в жир. А -
глкжагонпродуцирующие а-клетки поджелудочной
железы; В - инсулинпродуцирующие Р-клетки
поджелудочной железы
действием на обмен веществ. К инсулину может развиваться резистент-вость,острая резистентность связана с инфекциями или воспалением. Резистентность может определяться появлением в кровотоке антител к инсулину (IgG) и тканевой нечувствительностью, чточасто наблюдается при ожирении. Афинность (сродство рецепторов к инсулину) и/или число рецепторов зависит от ряда факторов; это суль-фонилмочевинные препараты, рН, цАМФ, физическая активность, характер и состав пищи, антителаи другие гормоны.
Глюкагон - полипептид, состоящий из 29 «минокислот, секретируется а-клетками островковподжелудочной железы, секреция снижается при повышении концентрации глюкозы в рови.В основном его эффекты противоположны действию инсулина. Глюкагон стимулирует гжикогенолиз в печени и глюконеогенез и спо-
собствует липолизу и кетогенезу. Совместные эффекты инсулина и глюкагона в поджелудочной железе и на обмен веществ в печени представлены на рис. 86.
Адреналин синтезируется в мозговом слое надпочечников, в печени он стимулирует глико-генолиз и глюконеогенез, в скелетной мускулатуре - гликогенолиз и липолиз, в жировой ткани усиливает липолиз. Гиперпродукция адреналина наблюдается при феохромоцитоме, при этом в крови может быть транзиторная гипергликемия.
Глюкокортикоиды вырабатываются корой надпочечников, усиливают глюконеогенез, тормозят транспорт глюкозы, ингибируют гликолиз и пентозофосфатный цикл, снижают синтез белка, потенциируют действие глюкагона, катехо-ламинов, соматотропного гормона. Избыточной продукцией глюкокортикоида гидрокортизона
| Норма |
| г.'У;-; Тглюкоэы, Ткетоновых тел |
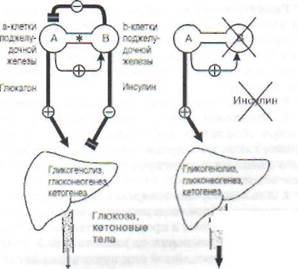 Недостаточный синтез инсулина
Недостаточный синтез инсулина
Рис. 86. Противоположные эффекты инсулина и глюкагона в поджелудочной железе и их действие на обмен веществ в печени. В норме печень образует примерно 10 г глюкозы в час, при этом 65-75% этого количества - глюкагонзависимо. Инсулин может угнетать секрецию глюкагона а-клетками независимо от уровня глюкозы крови. Инсулин обладает противоположным действием на печень, угнетая в ней образование глюкозы и кетоновых тел. При уменьшении отношения инсулин/глюкагон (при голодании) увеличивается образование глюкозы и кетоновых тел в печени. При инсулино-вой недостаточности в печени существенно повышается образование глюкозы и кетоновых тел
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
характеризуется синдром Иценко - Кушинга, при котором гипергликемия возникает из-за избыточного образования глюкозы из белков и других субстратов.
Гормоны щитовиднойжелезы усиливают скорость утилизации глюкозы, ускоряют ее всасывание в кишечнике, активируют инсулиназу, повышают основной обмен, в том числе окисление глюкозы. Тиреотропный гормон оказывает метаболические эффекты через стимуляцию щитовидной железы.
Соматотропный гормонобладает метаболическим эффектом, оказывает гипергликемичес-кое действие, в жировой ткани - липолитичес-кий эффект. При избытке образования СТГ у детей развивается гигантизм, у взрослых - акромегалия. Высокий уровень глюкозы в крови признак этого заболевания.
Адренокортикотропный гормонпрямо и через стимуляцию освобождения глюкокортикои-дов вызывает выраженный гипергликемический эффект.
Гипергликемия- повышение уровня глюкозы в крови выше 6,0 ммоль/л натощак. В норме концентрация глюкозы в крови натощак составляет 3,5 - 5,5 ммоль/л. Гипергликемические состояния у человека встречаются чаще, чем гипогликемии.
Различают следующие типы гипергликемий:
1. Физиологические гипергликемии.Это быстро обратимые состояния. Нормализация уровня глюкозы в крови происходит без каких-либо внешних корригирующих воздействий. К ним относятся:
1. Алиментарная гипергликемия. Обусловлена приемом пищи, содержащей углеводы. Концентрация глюкозы в крови нарастает вследствие ее быстрого всасывания из кишечника. У практически здоровых людей пик концентрации глюкозы в крови достигается примерно к концу первого часа после начала приема пищи и возвращается к верхней границе нормы к концу второго часа после еды. Механизм, обеспечивающий удаление избыточного количества глюкозы из кровотока, связан с эффектами инсулина: гормон, в частности, обеспечивает эффективный трансмембранный перенос молекул глюкозы из крови в цитоплазму клеток. Активация секреции гормона р-клетками островков Лангерганса поджелудочной железы начинается рефлектор-но, сразу после попадания пищи в полость рта и достигает максимума при продвижении пищи в
двенадцатиперстную кишку и тонкий кишечник. Пики концентраций инсулина и глюкозы в крови совпадают по времени. Таким образом, инсулин не только обеспечивает доступность углеводов пищи клеткам организма, но и ограничивает повышение концентрации глюкозы в крови, не допуская этим возможности потери ее с мочой.
2. Нейрогенная гипергликемия. Развивается в ответ на психологический стресс и обусловлена выбросом в кровь большого количества кате-холаминов (адреналина и норадреналина), образующихся в мозговом веществе надпочечников. Под влиянием повышенной концентрации кате-холаминов в крови происходит активация фермента аденилатциклазы, связанного с плазматическими мембранами клеток многих органов, прежде всего скелетных мышц и гепатоцитов. В результате этого в цитоплазме увеличивается концентрация циклического АМФ, под влиянием которого активируется фермент киназа фос-форилазы «Ь».Под действием упомянутой про-теинкиназы неактивная фосфорилаза «Ь» переходит в свою активную форму - фосфорилазу «а», которая является ключевым ферментом гли-когенолиза, определяющим скорость процесса распада гликогена в печени и мышцах. Освобождающаяся глюкоза быстро выходит в кровь, обусловливая гипергликемию. Физиологический смысл этого феномена состоит в обеспечении срочной мобилизации резерва углеводов для использования их в качестве источников энергии (окисления) в предстоящей повышенной мышечной работе.
2. Патологические гипергликемии.Их развитие может быть обусловлено:
1) нейроэндокринными расстройствами, в основе которых лежат нарушения оптимального соотношения между уровнями гормонов гипо- и гипергликемического действия в крови. Например, при заболеваниях гипофиза, опухолях коры надпочечника, при феохромоцитоме, гиперфункции щитовидной железы; при недостаточной продукции инсулина;
2) органическими поражениями центральной нервной системы, расстройствами мозгового кровообращения различной этиологии;
3) существенными нарушениями функций печени воспалительного или дегенеративного характера;
4) судорожными состояниями, когда происходит расщепление гликогена мышц и образо-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
вание лактата, из которого в печени синтезируется глюкоза;
5) действием некоторых видов наркотических веществ (морфин, эфир), возбуждающих симпатическую нервную систему и тем самым способствующих развитию гипергликемии.
Наиболее часто встречается гипергликемия при недостаточности инсулина - инсулинзави-симая гипергликемия, которая лежит в основе сахарного диабета.
Сахарный диабет
Сахарный диабет - это группа метаболических (обменных) заболеваний, характеризующихся гипергликемией, которая является результатом дефектов секреции инсулина, действия инсулина или обоих этих факторов.Хроническая гипергликемия при диабете сочетается сповреждением, дисфункцией и недостаточностью различных органов, особенно глаз, почек, нервов, сердца и кровеносных сосудов.
В развитие диабета вовлечены несколько патогенетических процессов: от аутоиммунного повреждения В -клеток поджелудочной железы с последующим дефицитом инсулина до нарушений, провоцирующих резистентность к действию инсулина. Основой нарушения метаболизма углеводов, жиров и белков при диабете является недостаточность действия инсулина в тканях-мишенях. Недостаток действия инсулина -результат неадекватной секреции инсулина и/ или сниженного тканевого ответа на инсулин в одной или нескольких точках на сложных путях действия гормона. Нарушение секреции инсулина и дефекты его действия часто сосуществуют у одного и того же больного, и порой неясно, какое нарушение является первичной причиной гипергликемии.
Симптомы выраженной гипергликемии включают полиурию, полидипсию, снижение массы, иногда с полифагией, и снижение остроты зрения. Ухудшение роста и восприимчивость к инфекциям также могут сопровождать хроническую гипергликемию. Острые, угрожающие жизни осложнения диабета - гипергликемия с кето-ацидозом, а также гиперосмолярный синдром без кетоза.
Хронические осложнения диабета включают ретинопатию с возможным развитием слепоты; нефропатию, ведущую к почечной недостаточности; периферическую нейропатию с риском образования язв на нижних конечностях и ам-
путации, а также сустава Шарко; автономную нейропатию, вызывающую гастроинтестиналь-ные, урогенитальные, сердечно-сосудистые симптомы и половую дисфункцию. Среди больных диабетом высока частота атеросклеротических поражений сосудов сердца, периферических и церебральных сосудов. Часто у больных обнаруживается гипертония, нарушения метаболизма липопротеидов и парадонтоз. Эмоциональное и социальное влияние диабета и потребности лечения могут вызвать существенную психосоциальную дисфункцию у больных и членов их семей.
Подавляющее большинство случаев диабета относится к двум обширным патогенетическим категориям. Причина диабета I типа (I категории) - абсолютный дефицит секреции инсулина. Лица с высоким риском развития этого типа диабета часто могут быть идентифицированы по серологическим признакам аутоиммунного патологического процесса в панкреатических островках, а также по генетическим маркерам. При диабете II типа (II категории, более распространенной) причина заключается в комбинации резистентности к инсулину и неадекватного компенсаторного инсулинсекреторного ответа. В этой категории степень гипергликемии достаточна, чтобы привести к патологическим и функциональным изменениям в органах-мишенях, но эта гипергликемия еще не вызывает клинических симптомов и может существовать достаточно долго до момента выявления диабета. В течение этого бессимптомного периода можно обнаружить нарушение углеводного обмена путем определения уровня глюкозы плазмы натощак или после пероральной нагрузки глюкозой.
Диабет I типа (деструкция (3-клеток, обычно ведущая к абсолютному дефициту инсулина)
Иммуноопосредованный диабет.Эта форма диабета обозначается также терминами: инсулин-зависимый сахарный диабет (ИЗСД), диабет I типа, диабет с ювенильным началом, - является результатом аутоиммунной деструкции В-клеток поджелудочной железы.
Маркеры иммунной деструкции (3-клеток включают аутоантитела к островковым клеткам (ICAs), аутоантитела к инсулину (IAAs), аутоантитела к декарбоксилазе глютаминовой кислоты (GAD65) и аутоантитела к тирозин-фосфата-
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
зам 1А-2 и IA2b. Один вид, а обычно более, этих аутоантител присутствует у 85-90% индивидуумов при первоначальном обнаружении гипергликемии натощак. Заболевание имеет четкую ассоциацию с HLA, связанную с генами DQA и В, а также на него влияют гены DRB. Эти аллели HLA-DR/DQ могут быть как предрасполагающими, так и протективными. Показано, что после вирусной инфекции резко возрастает частота проявления сахарного диабета I типа, это, в частности, характерно для вируса Коксаки В4, который относится к этиологическим факторам I типа СД. Воспаление островков поджелудочной железы (инсулиты) приводит к повреждению Р-клеток и снижению синтеза в них инсулина. Доказано, что Т-клеточная реакция, направленная против вирусных антигенов, может затрагивать клеточные антигены островков и тем самым вызывать повреждения клеток. Ведущую роль в развитии каскада иммунологических реакций, приводящих к деструкции р-клеток, отводят макрофагальным элементам, в том числе входящим в структуру островков Лангерганса. Под влиянием вирусов, химических агентов на поверхности р-клеток экспрессируется антиген. Макрофаги опознают этот антиген как чужеродный, вместе с Т-хелперами они выбрасывают интерлейкины и лимфокины (IL-1, у-интерферон, TNF), которые активируют иммунекомпетентные клетки. В результате этих процессов появляются аутоантитела к поверхностным и цитоплаз-матическим антигенам Р-клеток островков поджелудочной железы. Некоторые из антител используются в качестве маркеров СД I типа (JCA, GAD-65, GAD-67). Поскольку аутоиммунная деструкция протекает скрытно, то с момента запуска этих реакций до клинической манифестации (гибель 80-90% Р-клеток) проходит определенный период. Клинически возникновение СД I типа является конечным этапом процесса повреждения островковых клеток. При раннем обнаружении процесса поражения этих клеток и при адекватном лечении повреждение клеток можно остановить и предупредить. Показано, что рано начатое лечение иммуносупрессантом циклоспорином А может существенно снизить вероятность развития СД I типа после виремии.
При этой форме диабета прогрессирование деструкции р-клеток довольно различно, бывает быстрым у одних индивидуумов (главным образом у детей) или медленным (в основном у взрос-
лых). У некоторых, особенно у детей и подростков, в манифестации заболевания может быть представлен кетоацидоз. Другие имеют умеренную гипергликемию натощак, которая может быстро смениться выраженной гипергликемией и/или кетоацидозом при присоединении инфекций или стрессов. В то же время иные, особенно взрослые, могут сохранять остаточную функцию Р-клеток на уровне, достаточном для предотвращения кетоацидоза в течение продолжительного периода. Многие больные с такой формой диабета I типа в конечном счете становятся жизненно зависимыми от инсулина и находятся в состоянии риска по кетоацидозу. На этой последней стадии заболевания секреция инсулина мала или отсутствует, что проявляется низким или неопределяемым уровнем С-пептида плазмы. Иммуноопосредованный диабет обычно начинается в детском и подростковом возрасте, но может развиться в любой период жизни, даже у 80- или 90-летних стариков.
Аутоиммунная деструкция Р-клеток имеет множественные генетические предрасполагающие факторы, но на нее также влияют и факторы внешней среды, которые пока плохо изучены. Хотя больные редко имеют ожирение, его наличие не означает несовместимость с этим диагнозом. Пациенты с диабетом I типа также часто склонны к другим аутоиммунным заболеваниям, таким как болезнь Грейвса, тиреоидит Хашимото, болезнь Аддисона, витилиго и др.
Идиопатический диабет. Некоторые формы диабета I типа не имеют известной этиологии. Ряд таких больных имеют постоянную инсули-нопению и наклонность к кетоацидозу, но у них отсутствуют показатели аутоиммунного процесса. Хотя лишь меньшинство больных с диабетом I типа попадают в эту категорию, из тех, кто может быть к ней отнесен, большинство -африканского или азиатского происхождения. У пациентов с этой формой диабета эпизодически бывает кетоацидоз и представлены различные степени инсулинодефицита между такими эпизодами. Эта форма диабета имеет четкое наследование, недостаток данных по аутоиммунному поражению Р-клеток и не связана с HLA. Абсолютная потребность в заместительной инсули-нотерапии у этих больных может появляться и исчезать.
Актуальной задачей является диагностика предклинического периода развития деструкции
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
инсулярного аппарата. Это достигается с помощью определения аутоантител (JCA, GAD-65, GAD-67) или по оценке инсулинового ответа на внутривенную нагрузку глюкозой. Уже на стадии доклинических проявлений деструкции исчезает первая фаза инсулинового ответа на глю-козную нагрузку. Разработана так называемая модель двойных параметров (увеличение JCA + снижение I фазы секреции инсулина + антитела к инсулину). Если эти положительные признаки сочетаются с наследственной предрасположенностью (HLA типирование), то можно с большой вероятностью поставить диагноз деструкции инсулярного аппарата на доклинических этапах СД I типа.
Диабет II типа (от преобладающей инсулинрезистентности с относительным инсулинодефицитом до преобладающего дефекта секреции инсулина с инсулинрезистентностью)
Эта форма диабета определяется так же как инсулинонезависимый сахарный диабет (ИНСД), диабет II типа, диабет со «взрослым» началом -название, применяемое к больным, имеющим резистентность к инсулину и обычно относительную (чаще, чем абсолютную) недостаточность инсулина. Изначально, а часто на всем протяжении жизни, инсулин не является жизненно необходимым для этих больных. Возможно, существует много различных причин этой формы диабета, и, вероятно, доля больных этой категории в будущем уменьшится в результате идентификации специфических патогенетических процессов и генетических дефектов, которая сделает возможной их лучшую дифференциацию и более четкую субклассификацию.
Большинство больных с этой формой имеют ожирение, оно само по себе вызывает некоторую степень инсулинрезистентности. У больных, не имеющих ожирения по традиционным критериям массы, может быть повышенным процент жира тела, распределенного преимущественно в абдоминальной области. При этом типе диабета кетоацидоз редко развивается спонтанно, а когда наблюдается, обычно связан со стрессом в результате другого заболевания, например инфекции. Эта форма диабета часто остается неди-агностированной многие годы, так как гипергликемия развивается постепенно и ранние ста-
дии подчас недостаточно выражены, чтобы больной мог отметить какие-либо из классических симптомов диабета. Тем не менее такие больные находятся в состоянии повышенного риска мак-ро-и микрососудистых осложнений. Несмотря на то, что больные с этой формой диабета могут иметь уровни инсулина, представляющиеся нормальными или повышенными, можно было бы ожидать, что они были бы еще выше в ответ на высокую гликемию, если бы Р-клетки функционировали нормально. Таким образом, секреция инсулина у этих больных неполноценна и недостаточна для того, чтобы компенсировать инсу-линрезистентность. Резистентность к инсулину может уменьшиться в результате снижения массы и/или фармакотерапии гипергликемии, однако она редко восстанавливается до нормальной. Риск развития этого типа диабета возрастает с возрастом, ожирением и недостаточной физической активностью. Он возникает чаще у женщин с предшествовавшим сахарным диабетом беременных и у пациентов с гипертонией и дислипидемией, и его частота варьирует в разных расовых и этнических подгруппах. Некоторые характеристики сахарного диабета I и II типов представлены в табл. 38. Жизненно важно обращать внимание не только на обмен глюкозы, но и на ожирение, гипертонию, нарушение липидного обмена, курение, сердечно-сосудистую патологию, инфекции, побочные эффекты терапии.
До конца патогенез СД II типа неясен. Он ассоциирован с генетической предрасположенностью, в большей мере, чем аутоиммунная форма диабета I типа. Однако генетика этой формы диабета сложна и пока четко не определена. Конкордантность между двумя однояйцовыми близнецами составляет более 90% . Однако генетические факторы четко не определены. Несколько генов, которые могли бы иметь ключевое значение в развитии диабета, были идентифицированы, в том числе гены, ответственные за функцию гексокиназы и трансмембранный перенос глюкозы. Выявлено несколько генных ассоциаций, наличие которых увеличивает склонность к развитию СД II типа. Важны приобретенные признаки. Так, примерно у 40% больных имеет место ожирение. Клетки островков Лангерганса у больных СД II типа, как правило, гистологически нормальны, хотя у пожилых больных могут быть очаги амилоидоза. Это обычно скопле-
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
Основные признаки сахарного диабета I и II типов
Таблица 38
| Признак | 1 тип (ИЗСД) | II тип (ИНСД) |
| Частота заболеваемости, преобладание мужчин или женщин среди заболевших | 0,2-0,5%, оба пола поражаются одинаково | 2-4%, женщины болеют чаще мужчин |
| Возраст возникновения болезни | Дети, молодые люди | Взрослые люди, старики |
| Развитие симптомов | Острое | Постепенное |
| Телосложение | Худые | Часто ожирение |
| Потеря массы при заболевании | Как правило, происходит | Похудение очень редко |
| Запах кетонов изо рта | Бывает часто | Обычно запаха нет |
| Изменение состава мочи | Глюкоза и ацетон | Глюкоза |
| Концентрация инсулина в плазме | Низкая или не определяется | Часто нормальная, может быть повышенной |
| Антитела к островковым клеткам | Присутствуют | Отсутствуют |
| Наследственность | Поражено <10% родственников I степени родства, конкордантность среди идентичных близнецов 50% | Поражено >20% родственников I степени родства, конкордантность среди близнецов 90-100% |
| Ассоциация с HLA | В8, В15, Dw3, Dw4, DR3, DR4 | Нет ассоциации |
| Лечение (основное) | Инсулин | Диета, сульфанилмочевинные препараты |
ние белка амилина (состоит из 37 аминокислот), однако маловероятно, что амилин имеет значение в развитии СД II типа. У многих больных клетки островков, по-видимому, нечувствительны к глюкозе, поэтому у них нарушена секреция инсулина. У больных с ожирением часто концентрация инсулина в сыворотке выше нормы, что свидетельствует о резистентности периферических тканей к его действию.
Метаболический синдром X(инсулинрезис-тентность) составляет патофизиологическую основу сочетания СД II типа, гипертонической болезни, ишемической болезни сердца (ИБС). Основной причиной СД II типа является нечувствительность инсулинзависимых тканей (печень, мышцы, жировая ткань) к инсулину. Инсулин оказывает свое действие на клетки этих тканей путем связывания со специфическими рецепторами на клеточной мембране. При этом запускается ряд внутриклеточных процессов, направленных на захват глюкозы клеткой (фосфорилиро-вание белков-транспортеров) и внутриклеточный метаболизм глюкозы. Резистентность может проявляться на рецепторном и послерецепторном уровнях. При этом инсулин вначале продуцируется в нормальном или избыточном количестве.
Хроническая гипергликемия может привести к нарушению функции Р-клеток путем их истощения, поэтому предложен термин «глюкоток-сичность», который отражает влияние гипергликемии в развитии СД II типа.
Ранняя диагностика синдрома инсулинрези-стентности может помочь в профилактике и лечении СД II типа и его осложнений. Проводить обследование на инсулинрезистентность необходимо при наличии факторов риска у пациентов (гипертоническая болезнь, ИБС у родителей, ожирение, индекс массы тела более 30 кг/м2 поверхности тела, увеличение триглицеридов, снижение ЛПВП). В исследование на инсулин-резистентность, помимо проведения теста толерантности к глюкозе (ТТГ), включают определение инсулина и С-пептида натощак и через 2 ч после дачи 75 г глюкозы. Даже при нормальном ответе со стороны глюкозы в ТТГ существенное увеличение инсулина и С-пептида свидетельствует о наличии инсулинрезистентности.
Вторичный сахарный диабет.Диабет считается «первичным»при отсутствии известного этиологического фактора и «вторичным»в том случае, если гипергликемия связана с известной причиной. Вторичный диабет может быть
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
как I , так и II типа. Наиболее часто вторичным бывает СД II типа. Оба типа сахарного диабета возникают у пациентов, предрасположенных к заболеванию, при воздействии факторов риска. Основными причинами вторичного сахарногодиабета являются:
1.Нарушение функции поджелудочной железы: хронический панкреатит (алкогольный, тропический и т.д.), фиброз, рак.
2. Болезни печени: цирроз, хронический активный гепатит.
3. Болезни эндокринных желез: акромегалия, синдром Кушинга, тиреотоксикоз, глюкагонома, феохромацитома, синдром Конна, гиперандроге-немия.
4. Гемохроматозы.
5. Лекарственный - индуцированный корти-костероидами, оральными контрацептивами, тиазидовыми диуретиками и диазоксидом, ва-кором и другими препаратами, токсически действующими на поджелудочную железу, циклоспорином А, пентамидином.
6. Генетические нарушения.
Алкогольный панкреатитявляется наиболее
частой причиной хронического панкреатита в западных странах. Заболеванию подвергаются люди в основном среднего возраста. В диагностике помогают клинические стигмы хронического алкоголизма, наличие цирроза печени, признаки портальной гипертензии. Уровень глюкозы в крови весьма неустойчив в связи с нарушениями в диете, ухудшением всасывания и переваривания, снижением эндокринной функции поджелудочной железы; в течение небольших периодов времени могут возникать гинер- и гипогликемии; кетоацидоз - явление редкое. Лечение ферментами поджелудочной железы улучшает гликемический контроль.
Обширные поражения печени,такие как цирроз, снижают экстракцию инсулина печенью из портальной циркуляции, что приводит к периферической гиперинсулинемии и инсулиноре-зистентности. У предрасположенных пациентов на этом фоне возникает диабет. Нарушение толерантности к глюкозе и диабет средней тяжести отмечаются у 50-80% пациентов с установленным циррозом.
Повышенный уровень контринсулярных гормонов,особенно у предрасположенных лиц, снижает резервную функцию р-клеток и приводит к гипергликемии. Диагностика этих состоя-
ний обычно не вызывает затруднений в связи с классической симптоматикой и клинической картиной, обусловленной избыточной продукцией гормонов.
Гемохроматозсопровождается отложением железа в печени, поджелудочной железе, коже, половых органах. Подозрение на наличие гемо-хроматоза у больного сахарным диабетом должно возникнуть при сочетании бронзового оттенка кожи, гепатомегалии с аномальными функциональными печеночными тестами и импотенции. Эффективное лечение гемохроматоза флеботомией и железосвязывающими препаратами улучшает толерантность к глюкозе.
Некоторые лекарственные препараты(см. выше) могут нарушать толерантность к глюкозе, вызывая либо инсулинрезистентность, либо дисфункцию р-клеток.
Часть редких генетических аномалий(нарушение запасания гликогена, семейная гипер-липидемия, атаксия Фридрикса, миотоническая дистрофия и др.) может сопровождаться сахарным диабетом. Несмотря на разнообразие форм, к категории «вторичного» сахарного диабета клинически может быть отнесена относительно небольшая его часть (< 1%). Устранение или лечение основного заболевания у этих пациентов может привести к «излечению» сахарного диабета. «Вторичный» сахарный диабет развивается у лиц, предрасположенных к «первичному» диабету. По мере распознавания механизмов действия факторов риска первичного диабета, возможно, все большая часть случаев будет отнесена к категории вторичного.
Метаболические осложнения сахарного диабета
Патогенез диабетического кетоацидоза.При
сахарном диабете формируются нарушения всех видов обмена в организме: углеводного, белкового, липидного, электролитного и водного. Недостаток инсулина обусловливает выраженные дисфункции дыхательной, нервной и сердечнососудистой систем, почек и желудочно-кишечного тракта. Дефицит инсулина снимает ограничения в действиях его антагонистов: глюка-гона, катехоламинов, глюкокортикоидов и гормона роста. Патологические сдвиги метаболизма, вызываемые диабетом, по своей сути очень напоминают таковые, формирующиеся в усло-
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ 285
Таблица 39 Клинические и метаболические признаки диабетического кетоацидоза
 Клинические признаки Жажда, полиурия, дегидратация
Клинические признаки Жажда, полиурия, дегидратация
Гипотония, тахикардия, периферическая
циркуляторная недостаточность
Кетоз
Гипервентиляция
Тошнота, рвота
Абдоминальные боли
Затуманенное сознание, кома
Лабораторные показатели
Гипергликемия
Глюкозурия Метаболический ацидоз
Гипокапния
Кетонемия, кетонурия
Уремия
Гиперкалиемия
Гипертриглицеридемия
виях адаптации к непродолжительному голоданию практически здорового человека (лишение пищи в течение 3-5 сут, без ограничения в питьевой воде), но чрезвычайно сильно выражены (табл. 39). Ведущие патогенетические звенья диабетического ацидоза, учитывающие нарушения всех видов обмена, представлены на интегральной схеме 19.
Среди важнейших метаболических нарушений при сахарном диабете выделяются следующие:
1.Гипергликемия.Обусловлена нарушением поступления глюкозы из крови внутрь клеток, компенсаторным ускорением гликогенолиза, активацией глюконеогенеза вследствие снятия репрессивного действия инсулина (в условиях его дефицита) на синтез ключевых ферментов этого метаболического пути, а также благодаря усилению секреции глюкокортикоидов, являющихся стимуляторами синтеза ключевых ферментов глюконеогенеза в печени и почках.
Глюкозурия и полиурия.При достижении концентрации глюкозы в крови 10 ммоль/л преодолевается почечный барьер (нарушается способность почечных канальцев к реабсорбции глюкозы) и глюкоза появляется в моче. Вместе с ней организм теряет значительное количество воды, что обусловливает у больных диабетом постоянную жажду (полидипсия),по причине которой они могут выпивать до 20 л воды в сутки. Именно эти явления дали основания для исторически первоначального названия заболевания: diabetes mellitus (лат.) - сахарное мочеизнурение.
З.Кетонемия и кетоацидоз.Дефицит инсулина при сахарном диабете приводит к тому, что: а) контринсулярные гормоны (адреналин, глю-кагон, глюкокортикоиды и др.) стимулируют мобилизацию липидов из жировых депо и дос-
тавку жирных кислот к органам, что является адаптивным механизмом, поставляющим альтернативный субстрат окисления в условиях снижения утилизации глюкозы; б) начинает преобладать эффект глюкагона, стимулирующий ке-тогенез в печени; в) в норме кетоновые тела стимулируют выход инсулина из поджелудочной железы, что угнетает липолиз и, таким образом, ограничивает доставку липидов в печень и, соответственно, кетогенез. При сахарном диабете этот регуляторный механизм нарушен: идет усиленная продукция кетоновых тел печенью благодаря интенсивному Р-окислению жирных кислот; г) при сахарном диабете в избыточном количестве образуется продукт (3-окисления жирных кислот - ацетил-КоА, однако способность цикла Кребса утилизировать этот продукт существенно снижена. В результате этого избыток ацетил-КоА становится источником образования больших количеств кетоновых тел: р-оксимас-ляной, ацетоуксусной кислот и ацетона. Они начинают выделяться с мочой в виде натриевых солей (кетонурия),а ацетон - также и в составе выдыхаемого воздуха.
4.Нарушение кислотно-щелочного баланса организма.Развивается в связи с накоплением кислых продуктов метаболизма - кетоацидоз. По мере истощения емкости естественных буферных систем организма формируется некомпенсированный ацидоз.
5.Отрицательный азотистый баланс.Усиливается глюконеогенез из глюкогенных аминокислот, что приводит к дефициту в тканях пула свободных аминокислот и нарушению процесса синтеза белка. Стимулируется синтез мочевины.
б.Гиперосмотическая дегидратация тканей. Обусловлена потерей с мочой большого количества различных гидратированных соединений:
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
Схема 19 Патогенездиабетического кетоацидоза*
*Верхний ряд показывает основные метаболические эффекты при недостатке инсулина, которые вызывают все последующие изменения.
глюкозы, кетоновых тел, азотсодержащих соединений, ионов натрия, калия, хлора, неорганического фосфата и др.
Особо следует выделить нарушения обмена веществ, возникновение которых обусловлено именно диабетическим кетоацидозом. Так, у диабетиков формируется резистентность тканей к инсулину. Ацидоз (увеличение концентрации ионов водорода) препятствует реализации регу-ляторных эффектов инсулина вследствие нарушения гормон-рецепторного взаимодействия на поверхности плазматических мембран клеток-мишеней.
Сочетание ацидоза и явлений дегидратации в эритроцитах приводит к снижению в этих клетках концентрации 2,3-дифосфоглицериновой кислоты- аллостерического модулятора функций гемоглобина. В этих условиях сродство гемоглобина к кислороду возрастает, но его способность отдавать кислород тканям уменьшается, вследствие чего они получают меньше кис-
лорода. Это становится фактором, усугубляющим кислородное голодание тканей, которое уже сформировалось в ответ на недостаточность периферического кровообращения.
Диабетическая кома.Критическая дегидратация тканей организма с поражением функций головного мозга ведет к развитию диабетической (гипергликемической) комы.Кома развивается при достижении концентрации глюкозы в крови 22,0 ммоль/л и более. В этих условиях вследствие кетоацидоза ионы калия выходят во внеклеточное пространство (гиперкалиемия), что лежит в основе нарушения сократительной функции миокарда, а также дыхательной мускулатуры. Диабетическая кома может привести к летальному исходу, если больному не будет проведена специфическая противокоматозная терапия или же она проведена несвоевременно.
Различают следующие виды диабетической комы:
Дата добавления: 2015-03-19; просмотров: 2218;