Несовершенный остеогенез
Общие проявления. Термином «несовершенный остеогенез» обозначают наследственные аномалии, обусловливающие хрупкость костей (рис. 319-3). Диагноз уста
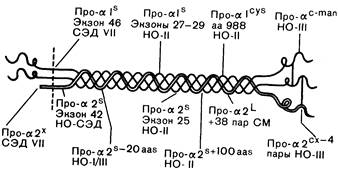
Рис. 319-2. Приблизительная локализация мутаций в структуре проколлагена I типа.
' Римскими цифрами обозначен конкретный тип синдрома Элерса—Данло (СЭД) или несовершенного остеогенеза (НО), обсуждаемых в тексте. Экзоны, в которых происходят специфические делеции, пронумерованы в направлении от 3'- к 5'-концу гена. Другие делеции обозначены примерным числом утраченных аминокислот; «аа 988» означает, что остаток глицина в положении 988 1-цепи замещен цистеином. Как сообщалось в тексте, мутация про-21 означает вставку 38 пар оснований в дополнительную последовательность и обнаружена у больных с атипичным синдромом Марфана (СМ); про-2^looaas означает делецию примерно 100 аминокислот при -варианте несовершенного остеогенеза II типа.
Про-^—мутация, ведущая к укорочению npo-al-цепи; про-(^—мутация, ведущая к укорочению ^1ро-а2-цепи; про-а!^5—мутация, ведущая к появлению цистеинового остатка; пpo-a':~ma"—мутация, ведущая к избыточному содержанию маннозы в одной или обеих про-а-цепях; про-а2" — неизвестная структурная мутация, препятствующая расщеплению цепи N-протеиназой; про-а21'— мутация, ведущая к удлинению про-а2-цепи; про-с^0" — мутация, меняющая структуру С-концевого пропептида про-а2-цепи (модифицировано и воспроизведено с разрешения из Prockop and Kivirikko).
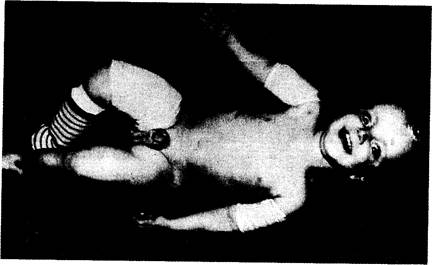
Рис. 319-3. Мальчик в возрасте 21 мес с несовершенным остеогенезом III типа. Ребенок страдает множественными переломами рук и ног. Он гомозиготен по делеции 4 пар оснований в генах про-а2(1)-цепей, что привело к изменению последовательности последних 33 аминокислот в этих белках. В связи с этим про-а2(1)-цепи не сомкнулись с про-а1 (I) -цепями, и единственной формой проколлагенов I типа оказались тримеры про-al (I) -цепей, в которых С-концевые участки остались нескрученными (воспроизве- навливают путем исключения других наследственных дефектов или влияний факторов окружающей среды, вызывающих остеопению или остеопороз, и выявления последствий мутации в нескольких видах соединительной ткани. Повышенная ломкость костей сопровождается обычно такими признаками, как голубой цвет склер, глухота, нарушение прорезывания зубов. Эти признаки могут определяться по отдельности или вместе (табл. 319-2). Для того чтобы установить диагноз в раннем детстве, достаточно выявить сочетание голубого цвета склер и переломов. Точно так же достаточно определить сочетание переломов с характерными аномалиями зубов (несовершенный дентиногенез). Некоторые специалисты диагностическое значение придают сочетанию ломкости костей с наступившей рано глухотой у больного или членов его семьи, тогда как другие ставят диагноз только на основании хрупкости костей, которую нельзя связать с внешними факторами (такие, как малая физическая активность или сниженное питание) или с другими наследственными синдромами, например дисплазиями скелета (табл. 319-3). Поскольку у некоторых членов семей переломов не бывает до наступления постменопаузы, легкие формы болезни могут быть неотличимы от постменопаузального остеопороза. Некоторые лица с остеопорозом могут быть гетерозиготными носителями генных дефектов, вызывающих у гомозигот несовершенный остеогенез. В связи с этим целесообразно отнести постменопаузальный остеопороз в спектр тех же болезней, к которым относится несовершенный остеогенез.
Для классификации несовершенного остеогенеза пользуются классификацией, предложенной Sillence (см. табл. 319-2). Тип I встречается с частотой примерно 1:30 000. Он представляет собой легкую или средней тяжести болезнь, наследуемую как аутосомный доминантный признак в сочетании с голубыми склерами. Наиболее тяжело протекает болезнь II типа. Типы III и IV по тяжести занимают промежуточное положение между типами I и II.
Аномалии скелета. При I типе болезни ломкость костей может быть столы выраженной, что ограничивает физическую активность больного, или столь незначительной, что больной вообще не испытывает никаких неудобств. При II типе кости и другие виды соединительной ткани настолько хрупки, что смерть наступает еще в утробном периоде, в родах или в первые несколько недель после рождения ребенка. При болезни III и IV типов множественные переломы, возникающие даже при минимальных физических воздействиях, могут привести к остановке роста и костным уродствам. У многих больных переломы особенно часто возникают в детстве; после периода пубертата их частота уменьшается, а при беременности и после наступления менопаузы вновь увеличивается. Резкий кифосколиоз может быть причиной нарушений дыхания и предрасполагать к легочным инфекциям. Плотность костей снижена, но относительно специфических морфологических нарушений мнения расходятся. Общее впечатление таково, что заживление переломов происходит нормально. У некоторых больных со сравнительно легкой симптоматикой череп имеет множество вмятин, по-видимому, из-за небольших очажков оссификации.
Таблица 319-2. Классификация несовершенного остеогенеза, основанная на клинических проявлениях и способе наследования (по Sillence)
| Тип | Ломкость костей | Голубые склеры | Аномалии зубов | Глухота | Наследование |
| I | Легкая степень | Определяются | Отсутствуют при IA, определяются при 1Б | В некоторых случаях | АД |
| II | Резко выраженная | То же | В некоторых случаях | Неизвестно | AD или f Л. Г НЛп ^- |
| III | Выраженная | Голубоватый цвет при рождении | То же | Редко | AD Л.Г |
| IV | Вариабельная | Не определяются | Отсутствуют при IVA, выявляются при ГУБ | То же | АД |
Примечание. АД — аутосомное доминантное; АР — аутосомное рецессивное; С — спорадическое.
Таблица 319-3. Частичная дифференциальная диагностика несовершенного остеогенеза
| Возраст При рождении | Диагноз Гипофосфатазия | Отличительные признаки Отсутствие минерализации костей черепа |
| Ахондрогенез | Отсутствие минерализации позвонков | |
| Танатоформная карликовость Вызывающая асфиксию дистрофия грудной клетки | Н-образные позвонки Цилиндрическая форма грудной клетки | |
| Ахондроплазия | Крупная голова, короткие трубчатые кости | |
| Младенчество | Синдром детских ушибов | Чаще переломы костей черепа и ребер |
| Цинга Врожденный сифилис | ||
| Детство | Идиопатический ювенильный остеогенез | В препубертатный период, спонтанно купирующийся |
| Гомоцистинурия | Марфаноидный внешний вид и отсталость психического развития | |
| Детская диарея Опухоль коры надпочечников Лечение кортикостероидами | Стеаторея, анемия |
Источник: модифицировано из Smith et al., с. 126.
Глазные симптомы. Цвет склер варьирует от нормального до слегка голубоватого или от синевато-серого до ярко-голубого. Голубизна обусловлена истончением или прозрачностью коллагеновых волокон склеры, через которые просвечивает сосудистая оболочка глаза. У ряда больных выявляют и другие глазные симптомы. В некоторых семьях голубые склеры могут быть наследственным признаком без всякого увеличения хрупкости костей.
Несовершенный дентиногенез. Эмаль твердой зубной пластинки относительно нормальна, но зубы имеют янтарный, желтовато-коричневый или полупрозрачный голубовато-серый цвет из-за неправильного отложения дентина. Молочные зубы обычно мельче нормальных, а постоянные заострены и как бы имеют основание. Точно такие же аномалии зубов могут наследоваться независимо от несовершенного остеогенеза.
Глухота. В возрасте после 10 лет или позднее развивается глухота. Она обусловлена нарушением прохождения колебаний через среднее ухо на уровне основания стремени. При гистологическом исследовании обнаруживают недостаточную оссификацию, персистенцию хрящевых участков, которые в норме оссифицируются, и полоски скопления кальция.
Сопутствующие проявления. У многих больных и у членов многих семей выявляют аномалии и в других видах соединительной ткани. В некоторых случаях отмечают изменения кожи и суставов, неотличимые , от таковых при синдроме Элерса—Данло (см. далее). У небольшого числа больных выявляют нарушение функции сердечно-сосудистой системы, например регургитацию аортальных клапанов, пролабирование митральных, митральную недостаточность и хрупкость стенок крупных кровеносных сосудов. Могут иметь место гиперметаболизм с повышением уровня тироксина в сыворотке, гипертермия и чрезмерная потливость. При легких формах болезни сопутствующие симптомы могут выступать на первый план.
Способ наследования. Тип I болезни наследуется как аутосомный доминантный признак с непостоянной экспрессией, так что он может проявляться через поколение. При летальном варианте II типа наследование может быть аутосомным рецессивным, но в нескольких случаях II типа с выясненным генетическим дефектом имелись новые мутации. Способ наследования — основной критерий разграничения III и IV типов (см. табл. 319-2), но отличить рецессивно наследуемую форму от новой аутосомной доминантной мутации иногда очень трудно.
Молекулярные дефекты. Поскольку большинство тканей при несовершенном остеогенезе богато коллагеном I типа, считают, что многие его формы связаны с мутациями структурных генов этого белка, генов, определяющих его посттрансляционный процессинг, или генов, регулирующих его экспрессию. В настоящее время выяснены мутации генов проколлагена I типа при четырех вариантах II типа несовершенного остеогенеза. Один вариант характеризовался делецией в одном из аллелей гена про-al (I) (рис. 319-4). Она распространялась на три экзона, но не препятствовала транскрипции гена. В результате про-al (I)-цепь оказалась на 84 аминокислоты короче, чем в норме. Эта мутация была летальной, поскольку укороченная про-al (I)-цепь связывалась с нормальной про-al (I)- и про-а2(1)-цепями (см. рис. 319-4). Укорочение про-al (I)-цепи препятствовало скручиванию молекул в тройную спираль. В связи с этим большая часть проколлагеновых молекул оставалась нескрученной и быстро распадалась в процессе, называемом самоубийством белка, или негативной комплементарностью (см. рис. 319-4). При втором летальном варианте болезни II типа мутация привела к синтезу такой про-а2(1)-цепи, которая была примерно на 20 аминокислот короче по сравнению с нормой. Второй аллель не функционировал, поэтому все про-а2-цепи оказались укороченными. При третьем варианте II типа мутационная делеция в аллеле про-а2(1)-цепи укоротила синтезируемую про-а2-цепь примерно на 100 аминокислот. При четвертом варианте II типа происходило замещение одного основания, что привело к появлению в a 1(1)-цепи остатка цистеина вместо глицина и тем самым к разрыву трехспиральной конформации белка.
Мутации генов проколлагена I типа выяснены также при двух вариантах болезни III типа. При одном из них была определена делеция четырех пар оснований, что изменило последовательность последних 33 аминокислот в про-а2(1)-цепи. Больной был гомозиготен по этому дефекту, и ни одна из про-а2(1)-цепей не включалась в молекулы проколлагена. Вместо этого проколлаген I типа состоял из тримера про-al (I)-цепей. Этот тример имел трехспиральную конфигурацию, но был нестабильным. Родители больного, находившиеся друг с другом в троюродном родстве, были гетерозиготами по той же мутации и уже в возрасте 30 лет страдали остеопорозом. При другом варианте III типа структурные изменения в С-концевом пропептиде обусловили увеличение количества в нем маннозы. У больного с некоторыми симптомами болезни I типа и другими, типичными для болезни II типа, про-а2(1)-цепи были укорочены примерно на 100 аминокислот.
На основании этих данных можно сделать ряд обобщений в отношении мутаций генов коллагена. Одно из них сводится к тому, что мутация, ведущая к синтезу аномального белка, может быть более вредной, чем нефункционирующий аллель. Второе заключается в том, что мутации, обусловливающие укорочение полипептидных цепей, могут быть более частыми, чем другие. Однако у большинства больных молекулярные дефекты не идентифицированы. У многих из них могли иметь место мутации сплайсинга РНК или мутации по единичным основаниям, которые трудно обнаружить в столь крупных генах, как ген проколлагена I типа. Ряд вариантов несовершенного остеогенеза мог бы обусловливаться мутациями других генов, экспрессия которых необходима для сборки и сохранения структуры костей и других видов соединительной ткани.
Диагностика. В отсутствие кардинальных признаков болезни диагноз установить трудно, и многие случаи, вероятно, остаются недиагностированными. Следует учитывать возможность других патологических состояний, сопровождающихся хрупкостью костей в младенчестве и детстве (см. табл. 319-3). У 1/3 больных при электрофорезе проколлагена I типа (синтезируемый фибробластами кожи в культуре) в полиакриламидном геле можно обнаружить аномальную про--цепь. В большинстве случаев изменение подвижности отражает посттрансляционную модификацию и не позволяет определить точную природу мутации или тип болезни.
Лечение. Убедительные данные о возможности эффективного лечения отсутствуют. При легкой форме после уменьшения частоты переломов в возрасте 15— 20 лет больные могут и не нуждаться в лечении, но во время беременности или после наступления менопаузы, когда частота переломов снова увеличивается, к ним требуется особое внимание. При более тяжелых формах детям необходимы широкая программа физиотерапии, хирургическое лечение при переломах и. деформациях скелета, профессиональное обучение и эмоциональная поддержка как больному, так и его родителям. У многих больных интеллект достаточно развит, и они, несмотря на выраженные деформации, делают успешную карьеру. Целесообразно использовать программу поддержания позы, разработанную Bleck. При многих переломах лишь минимально смещаются кости и происходит некоторый отек мягких тканей, поэтому требуется лишь слабое вытяжение в течение 1—2 нед с последующим наложением легкой шины. При малоболезненных переломах необходимо рано начинать физиотерапию. В отношении целесообразности коррекции деформаций конечностей с помощью стального гвоздя, помещаемого в длинные кости, мнения противоречивы. Оправданием этой процедуры может служить то обстоятельство, что коррекция деформаций в детстве дает возможность взрослым больным нормально ходить.
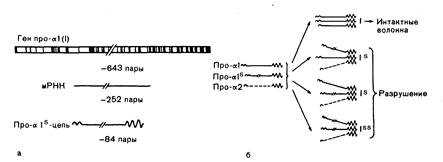
Рис. 319-4. Схематическое изображение молекулярного дефекта при несовершенном остеогенезе II типа. а: схематическое изображение генной делеции. Как упоминалось в тексте, у человека ген про-а1(1) состоит из 18000 пар оснований и содержит около 50 экзонов (вертикальные темные черточки). Делеция захватила три экзона, содержащих 252 пары оснований кодирующих последовательностей, б: схема «самоубийства белка», или негативной комплементарности. Синтезированные укороченные про-al (1)-цепи соединились и связались дисульфидными мостиками с интактными npo-a(I)-цепями. Молекулы проколлагена, содержащие одну или две укороченные про-al (I)-цепи, не скручивались в тройную спираль при 37 °С и разрушались. В результате при спорадическом гомозиготном дефекте количество функционирующего проколлагена было уменьшено примерно на 75 % (модифицировано и воспроизведено с разрешения из Prockop and Kivirikko).
Генетическое консультирование при II, III и IV типах болезни затруднено из-за неясности способа наследования. С помощью рентгене- и эхографии несовершенный остеогенез удавалось диагностировать у плода уже на 20-й неделе беременности. В тех немногих семьях, где точно выяснен генный дефект, для пренатальной диагностики можно было бы производить анализ ДНК в соответствующих лабораториях. Для генов проколлагена I типа идентифицирован полиморфизм длины рестрикционных фрагментов, и этот подход можно было бы использовать для пренатальной диагностики. Культура клеток амниотической жидкости синтезирует коллаген, но применять эти культуры для выявления мутаций представляется нереальным.
Дата добавления: 2015-03-17; просмотров: 1546;