Ретинобластома
По данным Европейской ассоциации офтальмологов в последние годы частота ее в популяции составляет 1 на 10 000-13 000 новорожденных. Выделены две формы заболевания: наследственная и спорадическая. У 10% больных ретинобластома сопровождается хромосомной патологией, в остальных случаях – структурными и функциональными нарушениям в гене RB1, который в последние годы был выделен и клонирован, благодаря использованию молекулярных маркеров. Белковый продукт этого гена функционирует в нормальных тканях и других опухолях, и только в случаях ретинобластомы он оказывается измененным. Таким образом, предрасположение к возникновению ретинобластомы в настоящее время связывают с наличием терминальной мутации в одной из аллелей гена RB1, которая передается по аутосомно-доминантному типу наследования и обнаруживается у 60-75% больных. Опухоль развивается у детей в раннем возрасте (до одного года). В 2/3случаев наследственная форма ретинобластомы оказывается билатеральной. Кроме того, при семейных формах ретинобластомы ген RB1 оказывается поврежденным во всех соматических клетках, поэтому риск появления опухолей других локализаций у таких больных высок (около 40%). Исследование точковых мутаций в гене ретинобластомы хромосомным анализом позволяет в настоящее время подтвердить или исключить наследственную форму этой опухоли не только в семьях, отягощенных по ретинобластоме, но объяснить наследственную форму этой опухоли у детей от здоровых родителей. Обнаружение ретинобластомы у ребенка до 10 месячного возраста свидетельствует об ее врожденном характере, ретинобластому, симптомы которой появились после 30 месяцев, можно расценивать как спорадический случай.
Спорадическая форма составляет около 60% всех ретинобластом, всегда односторонняя, возникает после 12-30 месяцев жизни ребенка в результате мутаций de novo в обеих аллелях гена RB1, находящихся в клетках сетчатки.
Ретинобластома развивается из клеток эмбриональной сетчатки, ее относят к нейроэктодермальным новообразованиям с признаками эпендимальной и невральной дифференциации, характерны клеточные формирования в виде розеток Флекснера-Винтерштейнера. Отсутствие стромы способствует быстрому рассеиванию клеток опухоли с образованием сателлитов: при эндофитном росте – в стекловидное тело, в камеры глаза; при экзофитном – в субретинальное пространство, хориоидею, диск зрительного нерва и его межоболочечное пространство. Обнаруженный в ретинобластоме фактор васкулярного эндотелиального роста (Vascular endothelial growth factor-VEGF) принимает участие в формировании сосудистой системы опухоли. Низкий уровень VEGF приводит к гипоксии в опухолевых клетках, но фокальная гипоксия в свою очередь стимулирует продукцию этого фактора в РБ, что и обуславливает рост опухоли.
До 90-95% случаев ретинобластому диагностируют у детей до 5 лет, практически одинаково часто страдают мальчики и девочки. Опухоль развивается в любом отделе оптически деятельной части сетчатки, в начале своего роста выглядит как нарушение четкости рефлекса на глазном дне. Позднее появляется сероватый мутный плоский очаг с нечеткими контурами. Далее клиническая картина меняется в зависимости от особенностей роста ретинобластомы. Выделяют эндофитный, экзофитный и смешанный характер роста опухоли.
При эндофитной ретинобластоме опухоль начинается во внутренних слоях сетчатки и характеризуется ростом в стекловидное тело.
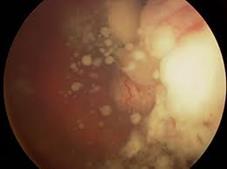
Эндофитный рост ретинобластомы
Поверхность опухоли бугристая. Толщина узла постепенно увеличивается, цвет сохраняется беловато-желтым, сосуды сетчатки и собственные сосуды опухоли не видны. В стекловидном теле над опухолью появляются конгломераты опухолевых клеток в виде «стеариновых капель», «стеариновых дорожек». Быстрый рост опухоли с нарушением в ней обменных процессов приводит к появлению некротических зон с творожистым распадом, впоследствии объизвествляющихся с формированием кальцификатов. Для эндофитной ретинобластомы характерно помутнение стекловидного тела за счет рассеивания опухолевых клеток. При локализации в преэкваториальной зоне клетки опухоли, оседая в задней и передней камерах глаза, создают картину псевдогипопиона, цвет которого в отличие от истинного, – беловато-серый. Рано появляется выворот зрачковой пигментной каймы. На поверхности радужки – узелки опухоли, массивные синехии, новообразованные сосуды. Передняя камера становится мельче, влага ее мутнеет. Увеличиваясь в размерах, опухоль заполняет всю полость глаза, разрушает и прорастает трабекулярный аппарат, в результате чего повышается внутриглазное давление. У детей раннего возраста развивается буфтальм, истончение склеролимбальной зоны, что облегчает распространение опухоли за пределы глаза. При прорастании опухоли склеры позади экватора развивается картина целлюлита, частота появления которого колеблется от 0,2% до 4,6% . Экзофитно растущая ретинобластома начинается в наружных слоях сетчатки и распространяется под сетчатку, что приводит к ее распространенной отслойке, купол которой удается видеть за прозрачным хрусталиком. Офтальмоскопически опухоль видна в виде одного или нескольких отграниченных узлов с ровной поверхностью.
В просвете зрачка серый узел опухоли с экзофитным ростом
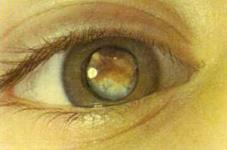
К опухоли подходят дренирующие расширенные и извитые сосуды сетчатки. На поверхности опухоли видны нежные извитые, хаотично расположенные новообразованные сосуды. Для ретинобластомы характерен мультифокальный рост. Сформированные узлы локализуются в разных участках глазного дна, имеют округлую или овальную форму, степень их толщины различная. Иногда геморрагии на поверхности опухоли сливаются, перекрывая по размерам диаметр опухоли. В подобных случаях при периферическом расположении ретинобластомы первым симптомом может оказаться «спонтанно» возникший гемофтальм. Смешанная ретинобластома характеризуется комбинацией офтальмологических симптомов, присущих описанным двум формам. Хорошо известные признаки, встречающиеся при ретинобластоме, – «свечение» зрачка и косоглазие, гетерохромия или рубеоз радужки, микрофтальм, буфтальм, гифема, гемофтальм – следует расценивать как косвенные, которые могут иметь место и при других заболеваниях. У 9,4% больных заболевание протекает без косвенных признаков и обнаруживается, как правило, при профилактических осмотрах.

«Свечение» зрачка при ретинобластоме
Ретинобластома у детей старшего возраста характеризуется снижением остроты зрения. В клинической картине преобладают признаки вялотекущего увеита, вторичной болящей глаукомы, отслойки сетчатки, редко ангиоматоза сетчатки. Возраст больных, когда вероятность ретинобластомы мала, усложняет правильную диагностику.
Трилатеральнуюретинобластому расценивают как билатеральную опухоль, сочетающуюся с эктопической (но не метастатической!) интракраниальной опухолью примитивного нейроэктодермального происхождения (пинеалобластомой).
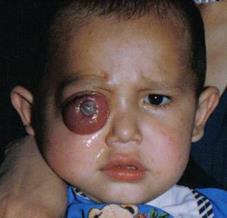
Трилатеральная ретинобластома
Третья опухоль локализуется, как правило, в области шишковидной железы, но может занимать и срединные структуры мозга. Клинически опухоль проявляется спустя 2-3 года после обнаружения билатеральной ретинобластомы признаками интракраниального новообразования. Трилатеральная ретинобластома выявляется у детей первых 4-х лет жизни. У маленьких детей признаки внутричерепного поражения могут проявить себя до появления видимых признаков поражения глаз.
В связи с неполной мутацией гена ретинобластомы, ретиноцитому расценивают, как редкий вариант ретинобластомы с более доброкачественным течением. Опухоль имеет лучший прогноз за счет наличия четких признаков дифференциации в виде формирования истинных розеток и склонности к самопроизвольной регрессии. Для диагностики ретинобластомы используют офтальмоскопию, которую следует проводить при максимальном расширении зрачка.У маленьких детей – в условиях медикаментозного сна ребенка. При осмотре глазного дна на крайней периферии необходимо использовать склерокомпрессию, что позволяет более детально осмотреть глазное дно в этих трудно доступных для визуального контроля участках. Офтальмоскопировать следует по всем меридианам (!). В затруднительных случаях при преэкваториальном расположении опухоли или наличии псевдогипопиона показана тонкоигольная аспирационная биопсия. Ультразвуковое сканирование дополняет диагностику ретинобластомы, позволяет определить ее размеры, подтвердить или исключить наличие кальцификатов. Компьютерная томография орбит и головного мозга показана детям старше 1 года жизни.
Лечениеретинобластомы комплексное, направленное на сохранение жизни больного ребенка и его глаза. Энуклеация, которую при ретинобластоме используют более 4-х веков, остается тяжелой ликвидационной операцией, не только инвалидизирующей детей, но и способствует появлению у них комплекса неполноценности со всеми вытекающими отсюда психологическими отклонениями. И не случайно частота энуклеации с конца 60-х годов 20 века при ретинобластоме снизилась с 96% до 75%. Увеличение частоты билатеральных форм ретинобластомы, стремление сохранить лучший глаз способствовали развитию органосохранного направления в лечении, которое включает в себя криодеструкцию, лазеркоагуляцию и лучевую терапию. Лечение ретинобластомы всегда индивидуально, планируется в зависимости от стадии процесса, общего состояния ребенка, фактора риска возникновения вторых злокачественных опухолей и ультимативного требования родителей сохранить зрение. При маленьких опухолях применение методов локального разрушения позволяет сохранить глаз в 83% случаев, а в комбинации с полихимиотерапией добиться 5-летней выживаемости почти 90% больных. Использование полихимиотерапии при больших опухолях в комбинации с энуклеацией способствует 4-летней переживаемости более чем у 90% больных. Ретинобластома диссеминирует вдоль зрительного нерва по межоболочечным пространствам, гематогенным путем распространяется в кости, головной мозг, лимфогенным путем в регионарные лимфоузлы. Прогноз для жизни при ретинобластоме зависит от ряда факторов: расположение опухоли кпереди от зубчатой линии, наличие множественных узлов опухоли, суммарный диаметр которых превышает 15 мм, объем опухоли, достигающий половины объема полости глаза и более, распространение опухоли в стекловидное тело или в орбиту, рост опухоли в хориоидею, зрительный нерв. Риск возникновения метастазов повышается до 78% при распространении опухоли в орбиту. Конечно, к фактору риска относятся и наследственные формы ретинобластомы. Стандартизированные показатели смертности от ретинобластомы при наследственных ее формах в последние годы увеличились с 2,9 до 9, в то время как при спорадических случаях ретинобластомы отмечено уменьшение их с 1,9 до 1,0.
С целью выявления ранних рецидивов опухоли после энуклеации или появлении опухоли в парном глазу обязателен контрольный осмотр ребенка. Его следует проводить каждые три месяца в течение 2-х лет при монолатеральной ретинобластоме, при билатеральной ретинобластоме – в течение 3-х лет. У детей старше 12 месяцев после окончания лечения один раз в год целесообразно проводить компьютерную томографию головы, что позволяет проконтролировать состояние мягких тканей орбит и исключить метастазы опухоли в головной мозг. Дети, излеченные от ретинобластомы, должны находиться под дисапнсерным наблюдением пожизненно.
Дата добавления: 2015-09-07; просмотров: 2408;