Настраиваем раздачу интернета по Wi-Fi с ноутбука на Windows 8 и Windows 8.1. Настройка точки доступа
| Особенности гемодинамики | Наличие цианоза | |
| нет | есть | |
| Обогащение малого круга кровообращения (МКК) | ДМЖП, ДМПП, ОАП, Аномальный дренаж легочных вен, неполная атриовентрикулярная коммуникация | Транспозиция магистральных сосудов (ТМС), ГЛС, общий артериальный ствол, единый желудочек сердца |
| Обеднение МКК | Изолированный стеноз легочной артерии (СЛА) | ТМС + СЛА, тетрада Фалло, трикуспидальная атрезия, болезнь Эбштейна, ложный ОАС |
| Препятствие кровотоку в БКК | Стеноз аорты (СА), коарктация аорты | |
| Без существенных нарушении гемодинамики | Декстрокардия, аномалия расположения сосудов, сосудистое кольцо — ДДА, болезнь Толочинова — Роже. |
Дефекты межпредсердной перегородки (ДМПП).
Дефекты межпредсердной перегородки — это группа ВПС, для которых характерно наличие аномального сообщения между двумя предсердными камерами. ДМПП составляют неоднородную группу аномалии эмбрионального развития межпредсердной перегородки и эндокардиальных валиков. Они различаются по расположению дефекта (центральный, верхний, нижний, задний, передний), его размеру (от небольшого щелевидного отверстия, например, при незаращении овального отверстия, до полного отсутствия МПП — единое предсердие), и количеству дефектов (от одного-двух до множественных). Неодинаково локализуются дефекты и по отношению к устьям верхней и нижней полых вен: верхние дефекты располагаются у устья верхней полой вены, нижние дефекты — над устьем нижней полой вены, в го время как множественные дефекты часто расположены центрально (Бураковский В.И. и др., 1996). ДМПП нередко сочетается с аномальным впадением (дренажом) вен: левой верхней полой веной, впадающей в левое предсердие; с аномальным частичным дренажом правых легочных вен в правое предсердие и др. Эти особенности также способны изменять характер и степень гемодинамических нарушений (Банкл Г., 1980).
По эмбриологическому генезу, т. е. в зависимости от характера и степени недоразвития первичной и вторичной межпредсердных перегородок и эндокардиальных валиков, выделяют первичные, вторичные дефекты и полное отсутствие МПП (единственное, общее предсердие, трехкамерное сердце).
ПервичныеДМПП возникают в результате недоразвития первичной МПП и сохранения первичного сообщения между предсердиями. Они чаще (4:1) сочетаются с открытым общим атриовентрикулярным каналом и дефектами атриовентрикулярных клапанов. Первичный ДМПП — это, как правило, большой по размеру дефект 1/3-1/2 часть перегородки), локализующийся в нижней части перегородки. Нижний край дефекта не имеет перегородочной ткани и образован перегородкой между атриовентрикулярными клапанами.
ВторичныеДМПП возникают вследствие недоразвития вторичной МПП, поэтому обычно полностью окружены ободком септальной ткани и в нижнем отделе всегда отделены краем МПП от перегородки, расположенной между двумя атриовентрикулярными клапанами. Размеры дефекта варьируются в достаточно широких пределах — от 2—5 до 20—30 мм в диаметре. В большинстве случаев дефект находится в центре МПП (65—67%); реже — в верхней (5-7%) и совсем редко — в задней (2,5%) и передней частях перегородки (Банкл Г., 1980; Бураковский В.И. и др., 1996).
Единственное (общее) предсердие формируется в результате недоразвития в эмбриональном периоде или полного отсутствия первичной и вторичной МПП и наличия большого дефекта, равного по площади всей МПП. При этом сохранены два предсердных ушка и дифференциальная структура правой и левой стенок предсердия. Поскольку нарушено развитие первичной МППи эндокардиальных валиков, то порок, как правило, сочетается с дефектом формирования атриовентрикулярных клапанов, а поэтому может рассматриваться как одна из форм ОАВК. При данном пороке нередко наблюдается аспления.
Относительно часто (15% случаев) ДМПП сочетается с другими врожденными аномалиями развития, например, с семейным синдромом Холта — Орама («сердце-конечность», предсердно-пальцевая дисплазия), который вероятно является следствием мутации гена, в результате которой нарушается нормальная одновременная дифференцировка сердца и верхних конечностей. Наиболее часто сочетается вторичный ДМПП с недоразвитием, а иногда и аплазией костей кисти, обычно левой (Holt M., Oram S., 1960). Отмечаются также семейные случаи ДМПП в сочетании с атриовентрикулярной блокадой (Porter J.C. et al, 1995).
Распространенность ДМПП колеблется в широком диапазоне — от 5% до 37,1%. Это, вероятно, обусловлено различным возрастным контингентом обследованных и сложностью раннего выявления и диагностики порока у детей младшего возраста. У взрослых ДМПП считается самым распространенным пороком, составляя 20—37% (Мешалкин Е.Н. и др., 1978; Минкин Р.Б., 1994), а у детей на его долю приходится 7,8—11%, он занимает второе-третье место по частоте встречаемости (Парийская Т.В., Гикавый В.И., 1989; Бураковский В.И. и др., 1996). ДМПП — это патология, распространенная преимущественно среди лиц женского пола (соотношение женщин и мужчин от 1,5 :1 до 3,5: 1) (Porter J.С. etal., 1995).
Естественное течениепорока и прогноз определяются размером дефекта и величиной артериовенозного сброса. Дети с вторичными ДМПП и малым сбросом крови развиваются нормально, не предъявляют жалоб, многие годы у них сохраняется физическая работоспособность, а первые симптомы неблагополучия иногда выявляются лишь в третьем десятилетии жизни. Однако в дальнейшем заболевание быстро прогрессирует, и большая часть пациентов умирают в возрасте до 40 лет, а живущие – к 50 годам становятся инвалидами (Банкл Г., 1980).
Младенческая смертность в основном обусловлена первичным ДМПП и (или) наличием ДМПП и АДЛВ, дефектов атриовентрикулярных клапанов и др., а также сочетанием ДМПП с другими экстракардиальными врожденными аномалиями. Непосредственными причинами смерти чаще всего являются тяжелые вирусные инфекции, рецидивирующие пневмонии, кишечные инфекции.
ДМПП реже, чем другие ВПС, осложняются инфекционным эндокардитом, хотя ревматизм у таких пациентов возникает относительно часто — в 10% случаев (Парийская Т.В., Гикавый В.И., 1989).
Дефекты межжелудочковой перегородки (ДМЖП).
Изолированный дефект межжелудочковой перегородки — это врожденное аномальное сообщение между двумя желудочками сердца, возникшее вследствие недоразвития МЖП на различных ее уровнях. Порок относится к наиболее частым ВПС у детей и встречается, по данным различных авторов (интернистов, хирургов, патологоанатомов) в 11—48% случаев (Парийская Т.В., Гикавый В.И., 1989; Бураковский В.И. и др., 1996; Банкл Г., 1980; Graham T.R, Gutgessell H.P., 1995 и др.).
МЖП в основном состоит из мышечной ткани и лишь в верхней части представлена небольшим участком фиброзной ткани в виде мембранозной (перепончатой) перегородки. Мышечная (средняя часть) преимущественно гладкая, а нижняя часть более грубая, трабекулярная. В соответствии с одноименными отделами правого желудочка МЖП разделяют на входную (приточную, в задней части перегородки), мышечную (трабекулярную, в средней и нижней части перегородки) и выходную (отточную, в передней и верхней части).
Дефекты МЖП могут возникать на границе, в месте стыка различных частей перегородки в результате их недоразвития. В области мембранозной части перегородки вследствие сохранения первичного межжелудочкового отверстия (в этом случае диаметр дефекта почти равен размеру мембранозной перегородки); в гладкомышечной и трабекулярной частях мышечной перегородки, когда все стороны дефекта образованы только мышечной тканью.
Существует множество классификаций ДМЖП, однако наиболее удобная и обоснованная — это классификация R.Anderson и J.Becker (1983), в которой учтены не только топографическое расположение дефектов, но и их связь с проводящей системой сердца и окружающими анатомическими структурами (атриовентрикулярными клапанами, клапанами аорты и легочной артерии).
Выделяют следующие дефекты МЖП:
1) приточный (типа АВК) перимембранозный;
2) приточный, субтрикуспидальный, трабекулярный, перимембранозный;
3) приточный, центральный, трабекулярный;
4) отточный, субаортальный, инфундибулярный, перимембранозный;
5) отточный, подлегочный, перимембранозный;
6) подаортально-подлегочный, инфундибулярный;
7) отточный, надгребешковый инфундибулярный;
8) верхушечный, трабекулярный;
9) отсутствие или рудиментарная МЖП.
Важной особенностью локализации ДМЖП является их соотношение с проводящей системой сердца. ДМЖП могут сочетаться с неполными и полными атриовентрикулярными блокадами, вследствие нарушения нормальной топографии атриовентрикулярного пучка Гиса, кроме того, проводящая система сердца может быть травмирована при хирургической коррекции порока.
При ДМЖП могут выявляться и другие сердечные аномалии: ДМПП (около 20% случаев); ОАП (20%); КоА (12%); СА (5%); врожденная недостаточность аортального клапана (2,5-4,5%); врожденная НМК (2%), крайне редко - СЛА, АДЛВ и др. (Банкл Г., 1980).
В 24—53% случаев ДМЖП сочетается с внесердечными аномалиями — болезнью Дауна (15%); дефектами конечностей (15%); пороками почек (8%); заячьей губой и расщелиной твердого неба (8%) (GrahamТ., Gutgessell H., 1995).
Течение и прогноз. ДМЖП относится к порокам, которые претерпевают значительную трансформацию в зависимости от величины и локализации дефекта и продолжительности заболевания.
Дефекты малых размеров, особенно расположенные в нижней мышечной части перегородки, имеют тенденцию к спонтанному закрытию. У 25—60% больных малые дефекты закрываются к 1—4 годам жизни, однако возможно спонтанное закрытие и в более старшем возрасте. Гораздо реже (примерно у 10% больных) происходит закрытие дефектов средних и даже больших размеров (Литасова Е.С., 1983; Бураковский В.И. и др., 1996; Банкл Г., 1980). Закрытие дефекта в мышечной, трабекулярной части перегородки происходит за счет роста окружающей дефект мышечной ткани, закрывающей дефект во время систолы. Кроме того, по мере взросления ребенка дефект малой величины почти полностью относительно уменьшается, и его влияние на гемодинамику исчезает за счет роста и увеличения размеров камер сердца. Закрытие дефекта может произойти за счет прикрытия дефекта дополнительной тканью трикуспидального клапана, образования аневризмы мембранозной перегородки, развития фиброза краев дефекта, пролабирования одной из створок аортального клапана (Белоконь Н.А., Подзол ков В. П., 1991; Anderson R.H. et al., 1983). При средних и больших дефектах МЖП, протекающих с большим сбросом слева направо, и длительном течении порока неизбежно развивается синдром (реакция) Эйзенменгера (дефект межжелудочковой перегородки субаортальной локализации, расширение ствола легочной артерии, гипертензия в малом круге кровообращения).
Другие осложнения порока: сердечная недостаточность, рецидивирующие застойно-бактериальные пневмонии, дистрофия и отставание в физическом развитии, наслоение инфекционного эндокардита, нарушения ритма сердца и проводимости, тромбоэмболия.
При средних и больших дефектах 50—80% больных умирают в возрасте до 1 года, и большая часть — до 6-го месяца жизни. Основной причиной смерти является сердечная недостаточность, особенно на фоне наслоившейся застойно-бактериальной пневмонии. Бактериальный эндокардит, нарушения ритма сердца, тромбоэмболические осложнения становятся причиной смерти примерно 10% больных, чаще детей старшего возраста. Важно подчеркнуть, что даже при благоприятном течении порока с малой величиной дефекта или при его спонтанном закрытии дети должны постоянно находиться на диспансерном учете у кардиолога, поскольку у них в разные сроки жизни могут возникать осложнения со стороны проводящей системы сердца в виде нарушений сердечного ритма и проводимости, а также при неблагоприятных условиях у них чаще, чем у здоровых детей, развивается инфекционный эндокардит.
Если у ребенка раннего возраста со средним или большим дефектом появляются признаки недостаточности кровообращения, то его необходимо лечить в стационаре, с применением кардиотонических препаратов, мочегонных; ингибиторов ангиотензинпревращающего фермента, кардиотрофических и антиоксидантных препаратов. Если такая терапия эффективна и не возникает признаков легочной гипертензии, то желательно проводить ее до достижения ребенком наиболее «операбельного» возраста, т.е. 3 и более лет, поскольку у детей раннего возраста операционная летальность высокая (Бураковский В.И. и др., 1996; Graham T.P., Gutgessell H.P., 1995). В то же время, если операция выполнена у ребенка младше 2—3 лет, то лучше проходит послеоперационная редукция легочной гипертензии и гипертрофии миокарда. Оптимальным для операции является возраст 5—9 лет.
Показания к операции:
1) отсутствие тенденции к спонтанному закрытию малых дефектов к 3— 4 годам жизни;
2) появление признаков легочной гипертензии;
3) стойкая рефрактерная к терапии сердечная недостаточность;
4) значительное отставание ребенка в массе тела и в физическом развитии, нарастание тяжести анемии;
5) тяжело протекающие и плохо поддающиеся терапии рецидивирующие пневмонии и бронхиты;
6) осложнение ДМЖП инфекционным эндокардитом, рефрактерным к консервативной терапии.
Операция противопоказанапри склеротической стадии легочной гипертензии, когда давление в легочной артерии приближается к системному.
Госпитальная летальностьзависит от возраста больных, тяжести сопутствующих заболеваний и, в наибольшей степени, от выраженности легочной гипертензии. У детей младшего возраста операционная летальность составляет 6—8%, а старшего — 1-4% (Бураковский В.И. и др., 1996; Винокуров А.В. и др., 1999). При коррекции малых дефектов без легочной гипертензии летальность составляет 1%, а при больших дефектах с легочной гипертензией — повышается до 10% (Алекси-Месхишвили В.В. и др., 1986; Белоконь Н.А., Подзолков В.П., 1991). Летальность более высока при пластике множественных дефектов.
Отдаленные послеоперационные результаты у большинства больных хорошие, особенно у детей (около 85% по данным Н.М.Амосова, Я.К.Бендета, 1983), оперированных в младшем возрасте и при отсутствии или умеренной выраженности легочной гипертензии. У детей наступает полная компенсация, а их физическая активность почти соответствует возрасту, однако требуется проведение ступенчатой комплексной реабилитации в течение длительного периода (лечебная физкультура, массаж, общеукрепляющая и витаминотерапия и др.) для восстановления здоровья.
Даже при хороших результатах операции у больных иногда длительно выслушивается хотя и менее выраженный, но «остаточный» шум, связанный с неизбежным умеренным послеоперационным нарушением архитектоники внутренних структур сердца, из-за чего ток крови становится турбулентным. Электрокардиографические и рентгенологические изменения, характерные для ДМЖП, уменьшаются или исчезают в течение нескольких лет. Вышеперечисленные положения, а также возможность послеоперационного развития инфекционного эндокардита требуют постоянного амбулаторного диспансерного наблюдения за этой категорией прооперированных пациентов.
Открытый артериальный проток (ОАП).
Открытый артериальный проток — это наличие аномального сосудистого сообщения между аортой и легочной артерией. Порок может быть изолированным или сочетаться с другими сердечно-сосудистыми аномалиями. Применяемое прежде название «незаращенный боталлов проток» связывали с именем итальянского врача Leonardo Botalli (1530—1600), однако первые анатомические описания ОАП принадлежат, вероятно, Галену (130—200 гг.), а объяснение функционального значения протока для пре- и постнатального кровообращения — Гарвею (Бураковский В.И. и др., 1996; Затикян Е.П., 1996).
ОАП относится к порокам бледного типа с обогащением МКК и является одним из наиболее часто встречающихся ВПС. По патологоанатомическим данным порок обнаруживается в 3—9,8% случаев, а по клиническим — в 5-34% случаев всех ВПС (Банкл Г., 1980; Бураковский В.И. и др., 1996; Затикян Е.П., 1996; Brook M.M., Heymann M.A., 1995). Порок четко и существенно преобладает у лиц женского пола в соотношении 2—4 : 1 (Бойков Г.А., Парийская Т.В., 1984; Банкл Г., 1980).
В 5—10% случаев ОАП может сочетаться (в порядке убывающей частоты) с другими ВПС (ДМЖП, КоА, СЛА, СА, ДМПП, НМК). В 15% случаев ОАП сочетается с аномалиями скелета (деформация грудины, косолапость, сколиоз), умственной отсталостью, дефектами глаз (Банкл Г., 1980).
Артериальный проток является сосудистым каналом, отходящим от переднелатеральной стенки дуги аорты, дистальнее отхождения левой подключичной артерии, идущим в косом направлении кпереди и вниз и впадающим в легочную артерию в области ее бифуркации или вблизи места отхождения левой легочной артерии. Реже артериальный проток может впадать непосредственно в правую или левую ветви легочной артерии, возможно также наличие двух протоков, впадающих в правую и левую ветви легочной артерии.
Проток имеет форму цилиндра или усеченного конуса, с основанием у аортального конца, быть относительно длинным (25—30 мм) и узким (2—3 мм), извилистым или коротким (3—5 мм) и широким (до 20—25 мм). Артериальный проток, в отличие от магистральных сосудов эластического типа, является мышечным сосудом с мощной вагусной иннервацией, что обеспечивает способность к его эффективному сокращению в раннем неонатальном периоде (Затикян Е.П., 1996). В дальнейшем в протоке может произойти наслоение инфекционного эндокардита («боталлит»), развиться аневризма (веретенообразная, мешкообразная, расслаивающая) или кальциноз. Длинные и узкие протоки создают большее сопротивление кровотоку через них и лучше облитерируются, а через короткие и широкие протоки, как правило, происходит большой сброс крови и возникают тяжелые гемодинамические нарушения. При последнем морфологическом варианте проток облитерируется редко.
Течение, осложнения и прогноз.При естественном течении порока средняя продолжительность жизни больных составляет 20—25 лет и зависит от величины дефекта и возникающих осложнений. Наибольшая смертность отмечается в фазу первичной адаптации у детей первых месяцев жизни, особенно недоношенных, и у больных после 30-летнего возраста. Так, если в течение первых 20 лет естественная смертность составляет около 0,5% в год и в первые 20—30 лет жизни умирают около 20% больных, то после 30 лет ежегодная смертность увеличивается до 4% в год и к 45 годам умирают42% больных, а к 60 годам — 50— 70% (Банкл Г., 1980).
Основными причинами смерти являются сердечная недостаточность, легочная гипертензия или осложнения ОАП инфекционным эндокардитом, аневризмой аорты, реже — легочной артерии.
Сердечная недостаточность у детей чаще возникает в первые месяцы (до 50% больных) или во второй декаде жизни, протекает как тотальная или (у более старших детей) как преимущественно правожелудочковая. В старшем возрасте СН может утяжеляться различными нарушениями ритма сердца и проводимости, которые обычно формируются на фоне выраженной дилата-ции полостей сердца, особенно предсердий.
Легочная гипертензия отмечается у 24-35% больных, из них у 14% она достигает склеротической стадии (Бураковский В.И. и др., 1996; 3атикян Е.П., 1996; Brook M.M., Heyman M.A., 1995).
Инфекционный эндокардит как осложнение порока чаще протекает в виде эндартериита (боталлита) артерального протока — у 0,33—5% больных (Бухарин В.А., 1975) и, как правило, возникает при узких и длинных протоках. Вегетации обнаруживаются преимущественно со стороны легочной артерии, в месте наибольшего сужения протока или в области противоположной стенки легочной артерии, подвергающейся наибольшей травматизации ударной струей крови из аорты. Реже вегетации наблюдаются на створках клапанов сердца. Эндартериит протока может протекать латентно или подостро. При развернутой клинической картине инфекционного эндокардита (эндартериита) возникают тромбоэмболические осложнения, в основном в системе легочной артерии или в БКК.
Аневризматические осложнения порока встречаются редко и могут быть в виде веретенообразной, мешкообразной или расслаивающей аневризмы.
При естественном течении порока его спонтанное закрытие после 6—12 месяцев жизни, в отличие от септальных дефектов, происходит значительно реже (примерно 0,6% в год), а большие дефекты практически никогда не закрываются.
Лечение.Показанием к операции является факт установления наличия порока у ребенка после 6—12 мес. жизни, так как возможность спонтанного закрытия уже минимальна, а существование даже небольшого дефекта чревато возникновением вторичных осложнений. Оптимальным для операции считается возраст 2—5 лет, поскольку отдаленные результаты при ранней коррекции дефекта наиболее благоприятны. Однако если при большом артерио-венозном шунте у детей раннего или более старшего возраста развиваются рефрактерная сердечная недостаточность, рецидивирующие пневмонии, тяжелая дистрофия или прогрессирующая легочная гипертензия, то операция проводится в любом возрасте.
Оперативное лечение не противопоказано и при наличии текущего инфекционного эндокардита (эндартериита). В этом случае необходимо провести стационарно в течение 1 — 1,5 мес. курс мощной, целенаправленной, сочетанной терапии антибиотиками, иммунокорригирующей и общеукрепляющей терапии, с последующей хирургической коррекцией, поскольку устранение дефекта уже является эффективным фактором купирования септического процесса. Хотя исходы операций на фоне текущего инфекционного эндартериита безусловно менее благоприятные. После операции эта категория больных нуждается в стационарном лечении антибиотиками и иммунокорректорами, а также в длительном наблюдении и периодическом обследовании на предмет выявления скрытого, подострого инфекционного эндокардита.
В настоящее время существует несколько методов коррекции дефекта: терапевтическая (медикаментозная) облитерация протока у новорожденных ингибиторами простагландинов Е2 и J2; катетерная эндоваскулярная окклюзия; оперативная перевязка ОАП или его пересечение с ушиванием аортального и легочного концов протока.
Метод медикаментозной облитерации используется только в последние два десятилетия. Он основан на ингибиции индометацином эндогенных простагландинов Е2 и J2, которые являются сильными дуктодилататорами. Ингибиция этих биологически активных веществ приводит к спазму и облитерации протока. Однако метод эффективен при его применении у новорожденных первых двух недель жизни.
Индометацин назначают как энтерально, так и внутривенно, однако при энтеральном применении закрытие протока происходит лишь у 18—20% новорожденных, в то время как при внутривенном — в 88—90% случаев (Бураковский В.И. и др., 1996; Brook MM., Heymann M.A., 1995). Внутривенно препарат вводят из расчета 0,1—0,2 мг/кг массы 1—2 раза в сутки, в течение 1—3 дней, под эхокардиографическим контролем. Курсовая доза не должна превышать 0,6 мг/кг массы тела.
Данный метод не получил широкого применения, так как имеет некоторые существенные ограничения и противопоказания. Индометацин обладает антиагрегантными свойствами и снижает агрегационную способность тромбоцитов, что может привести к внутричерепным и желудочно-кишечным кровотечениям, гипонатриемии, транзиторным ренальным дисфункциям. Лечение индометацином не проводят при наличии почечной недостаточности, энтероколита, нарушениях свертывающей системы крови, билирубинемии (выше 0,1 г/л).
При катетерной эндоваскулярной окклюзии в проток со стороны аорты с помощью специальных катетеров вводят «пробку» из синтетической ткани (Алекян Б.Г. и др., 2000; Portsman N., 1974). Боталлоокклюзия противопоказана при очень широком или очень коротком ОАП, а также при аневризме протока или текущем «боталлите». Отдаленные результаты операции хорошие, однако она пока не нашла широкого применения в хирургической практике (Бураковский В.И. и др., 1996).
Наиболее широко применяется метод закрытия ОАП путем перевязки протока либо пересечения с ушиванием обоих концов. Метод хорошо разработан, сравнительно безопасен и дает хорошие отдаленные результаты у 97— 98% оперированных больных, особенно детей первых лет жизни, у которых не развилась легочная гипертензия (Амосов Н.М., Бендет Я.А., 1983; Бойков Г.А., Парийская Т.В., 1984; Brook M.M., Heymann M.A., 1995). Послеоперационная летальность у больных с неосложненным течением (без легочной гипертензии, аневризмы протока, инфекционного эндокардита) практически сведена к нулю. Однако с возрастанием легочной гипертензии летальность увеличивается до 2,3—3% и больше (Королев Б.А., Романов Э.М., 1984; Бураковский В.И. идр., 1996). В редких случаях при лигировании широких протоков происходит их реканализация.
С появлением и прогрессированием легочной гипертензии от I до IIIА гемодинамических групп эффективность операции снижается с 99,5% до 80% (Бураковский В.И. и др., 1996). В группах больных с высокой, склеротической легочной гипертензией (IIIБ), несмотря на успешную хирургическую коррекцию порока, у большинства пациентов сохраняется или прогрессирует легочная гипертензия. Поэтому противопоказанием к хирургической коррекции порока является высокая, склеротическая легочная гипертензия, особенно при наличии «смены шунта» (появление перекрестного или стойкого цианоза), когда заболевание становится «дуктусзависимым», а ОАП — «предохранительным клапаном» для сброса части крови в БКК, разгрузки МКК и облегчения работы правого желудочка.
Тетрада Фалло (ТФ).
Тетрада Фалло — многокомпонентный ВПС, включающий в себя стеноз выводного отдела правого желудочка, дефект межжелудочковой перегородки, декстропозицию атероматозно расширенной аорты и гипертрофию миокарда правого желудочка. Порок относится к ВПС цианотического типа с обеднением МКК. ТФ выявляется, по клиническим данным, у 11 — 13%, а по патологоанатомическим данным — у 15—16,7% всех больных с ВПС. При этом, ввиду особенностей ранних проявлений порока, он гораздо реже диагностируется у новорожденных (5—8%), чем у детей более старшего возраста (12—14%). Данный порок является самым распространенным (до 75%) из всех ВПС цианотического типа (Keith J. etal., 1978; Банкл Г., 1980; Parenzan L., 1999).
В 40% случаев ТФ сочетается с другими сердечными аномалиями, в частности в 15% наблюдений с ДМПП.Такой пятикомпонентный патологический комплекс называют пентадой Фалло. В 16—25% случаев выявляется правосторонняя дуга аорты, с расположением левой подключичной артерии в правой половине грудной клетки (Банкл Г., 1980). Реже встречаются сопутствующие ОАП или анастомоз между легочной и подключичной артериями, за счет которых осуществляется компенсация легочного кровообращения, а спонтанное закрытие ОАП может приводить к катастрофическому ухудшению состояния больных. Еще реже обнаруживаются левая верхняя полая вена, частичный АДЛВ, гипоплазия левых отделов сердца и др.
В 10—20% случаев при ТФ могут быть и другие внесердечные аномалии (синдром Нунана, синдром Гольденара, синдром «кошачьего глаза» и др.) без характерных комбинаций (Затикян Е.П., 1996; Банкл Г., 1980). Порок возникает почти с одинаковой частотой у лиц мужского и женского пола.
Патологическая анатомия.Согласно современным представлениям, в основе нарушения эмбриогенеза лежит ротация артериального конуса против часовой стрелки и смещение конусной перегородки вперед и влево. Это не позволяет соединиться аорте с МЖПи формирует дефект МЖП, а устье аорты оказывается расположенным над МЖП («сидящая верхом» над перегородкой аорта). Кроме того, смещение конусной перегородки вперед-влево приводит к сужению и обструкции выходного тракта правого желудочка.
Патологический сердечный комплекс тетрады Фалло, состоящий из 4 компонентов, определяет клинико-гемодинамическую ситуацию, течение порока и его тяжесть, которые зависят от характера и выраженности каждого из компонентов комплекса, и прежде всего — степени стенозирования выходного отдела правого желудочка.
1. Сужение выходного отдела (артериального конуса) правого желудочка, затрудняющего отток крови в легочную артерию, является главным, определяющим компонентом порока. Обструкция может быть расположена на разных анатомических уровнях: в инфундибулярном отделе правого желудочка, на уровне клапана легочной артерии, по ходу ствола и ветвей легочной артерии или на нескольких уровнях.
2. ДМЖП при ТФ, как правило, инфундибулярный, большой, мембранозный, располагается под корнем аорты, на заднем или нижнем крае наджелудочкового гребешка. Площадь дефекта может составлять от 80% до 150% площади устья аорты.
3. Декстрапозиция аорты при ТФ чаще является не истинной, а вторичной и рассматривается как смещение ее устья вправо по отношению к МЖП. Декстрапозиция обусловлена также и атероматозно расширенной аортой, устье которой нависает над ДМЖП и является ее верхним краем. При этом фактически аорта в значительной степени отходит не только от левого, но и от правого желудочка, а степень смещения ее вправо по отношению к оставшейся мышечной части МЖП у больных может колебаться от 30% до 50%. Если аорта более чем на 50% отходит от правого желудочка, то многие авторы относят такую форму порока к двойному отхождению аорты и легочного ствола от правого желудочка (Бураковский В.И. и др., 1996).
4. Гипертрофия миокарда правого желудочка не является истинным первичным врожденным дефектом, так как у плода не обнаруживается (Затикян Е.П., 1996; Lev M. et al., 1964). Гипертрофия формируется как компенсаторный механизм, действующий при систолической перегрузке правого желудочка сопротивлением. Однако выявление выраженной гипертрофии правого желудочка в ранние сроки, ее быстрое прогрессирование, а также постоянное обнаружение у всех страдающих болезнью Фалло позволили считать ее четвертым компонентом порока. Гипертрофия значительная и имеется уже в раннем возрасте. Толщина стенки правого желудочка может достигать 10—15 мм, с резко выраженной трабекулярностью строения миокарда и наличием очагового или диффузного утолщения эндокарда (Банкл Г., 1980). Резкая гипертрофия трабекул в выходном отделе правого желудочка может увеличивать его обструкцию.
При ТФ может также иметь место аномальное формирование и расположение больших коронарных стволов. Кроме того, при ТФ изменяется строение сосудов легких в виде атрофии эластических и мышечных волокон, а из-за разрастания интимы и тромбозов дистальных отделов легочной артерии происходит уменьшение просвета сосудов и ухудшается легочный кровоток (Белоконь Н.А., Подзолков В.П., 1991).
Течение, осложнения, прогноз.При естественном течении порока средняя продолжительность жизни зависит от выраженности стеноза легочной артерии и составляет в среднем 12—15 лет. Около 25% больных с тяжелой обструкцией выходного тракта правого желудочка умирают в течение первого года жизни, чаще в периоде новорожденности, 40—75% детей умирают до 2—3 лет, около 30% больных доживают до 10 лет, 10% — до 20, и лишь 1—5% больных доживают до 30-40 лет.
Причинами смерти в первые 10 лет жизни являются гипоксия, нарушения гемо- и ликвородинамики, тромбозы и абсцедирование сосудов головного мозга, у более старших детей и взрослых — инсульты, инфекционный эндокардит (5—10%), сердечная недостаточность (8% больных). Приблизительно у 1/3 больных развивается дистрофия и анемия, у 10% больных, на фоне нарушения мозгового кровообращения, возникают стойкие парезы, у 2—3% больных — туберкулез легких. Большая часть детей с цианотической формой ТФ к 5-7 годам отстают в умственном и интеллектуальном развитии от сверстников на 2—3 года. При интенсивных интеллектуальных нагрузках у них появляются головные боли, отмечаются умственная утомляемость, слабость.
У больных с ТФ иногда наблюдаются пролонгированные, немотивированные повышения температуры тела неинфекционного генеза, длительностью от нескольких недель до месяцев, что дает основание подозревать развитие инфекционного эндокардита. Этот неинфекционный фебрилитет обусловлен хронической гипоксией мозга (особенно чувствительного к гипоксии лимбико-ретикулярного комплекса), в результате которой нарушается работа центров терморегуляции.
Лечение (консервативная терапия). Вследствие анатомо-физиологических и гемодинамических особенностей порока и характера компенсаторных механизмов при ТФ, его консервативная терапия имеет специфику. Это касается, прежде всего, применения кардиотонических препаратов. Известно, что тахикардия, одышка и цианоз при ТФ обусловлены не сердечной недостаточностью, которая развивается редко, а аномальным поступлением венозной крови непосредственно в аорту, из-за наличия субаортального ДМЖП и «сидящей верхом» над ним аорты. Поэтому при назначении кардиотоников, увеличивающих сократимость не только левого, но и правого желудочка, вено-артериальный сброс будет увеличиваться. Кроме того, при усиленном сокращении правого желудочка возрастает фиброзно-мышечная спастическая обструкция его выходного отдела, что еще более усиливает сброс венозной крови в аорту. Клинический опыт также подтверждает, что сердечные гликозиды или синтетические катехоламины не улучшают состояние больных, а даже увеличивают цианоз и одышку. Однако возможно использование препаратов со слабым кардиотоническим эффектом (адонизид), оказывающих и седативное действие.
Учитывая то, что развивающаяся после 3—6 мес. релятивная или абсолютная анемия усугубляет артериальную гипоксемию и провоцирует развитие одышечно-цианотических приступов, необходимо раннее налаживание рационального питания с достаточным обеспечением белком, витаминами и микроэлементами (особенно железом, медью и кобальтом). Возникающая анемия чаще всего железодефицитная, поэтому назначают препараты железа с витаминами (ферроплекс, фесовит, иберет ликвид и др.), а более старшим детям — тардиферон, сочетая их с препаратами сульфата меди. Парентеральное введение препаратов железа (феррум-ЛЕК, ферковен и др.) назначают лишь при тяжелой анемии, с нарушением функции печени и ЖКТ.
При сгущении крови и высоком показателе гематокрита необходимо обеспечить адекватный питьевой режим (особенно в жаркое время года, при гипертермии, поносах и др.) не менее 1—1,5 л/сут или 100—150мл/кг/сут — за счет потребления соков (яблочного, апельсинового, сливового, гранатового, томатного), компотов и овощных отваров. В случаях остро нарастающей дегидратации необходимо и внутривенное введение жидкости в виде физиологических растворов или «поляризующей смеси».
Для улучшения реологических свойств крови и микроциркуляции назначают антиагрегантную и антикоагулянтную терапию — ацетилсалициловую кислоту, реже фенилин или курантил в малых дозах.
Наряду с применением антианемической и антикоагулянтной терапии необходим постоянный (2 раза в квартал) контроль показателей крови (эритроциты, гемоглобин, время кровотечения, свертываемость, количество тромбоцитов, содержание фибриногена, протромбиновый индекс).
Для профилактики гипоксических приступов, в противоположность сердечным гликозидам, назначают β-адреноблокаторы (индерал, обзидан, анаприлин, тразикор и др.), которые, уменьшая сократимость миокарда, предупреждают возникновение мышечного спазма при инфундибулярном стенозе. Кроме того, они уменьшают частоту сердечных сокращений, потребность миокарда в кислороде и возникновение нарушений сердечного ритма. Однако эти препараты следует использовать с осторожностью при атриовентрикулярных блокадах и артериальной гипотензии.
Для борьбы с гипоксией мозга и улучшения мозгового кровообращения и ликвородинамики назначают диакарб, кавинтон, ноотропные препараты и церебропротекторы (пирацетам, аминалон, энцефабол, глютаминовую кислоту и др.).
Для уменьшения гипотрофии и улучшения анаболических и обменных процессов назначают нестероидные (оротат калия, инозин, рибоксин, мега-L-карнитин) или стероидные (ретаболил, неробол) анаболики, поливитаминные препараты, витамины В12 и В15, антиоксиданты (витамины А, С, Е, селен).
Показания к операции.Операция показана всем больным с ТФ. На первом году жизни необходимость в неотложной госпитализации возникает:
1) при крайней форме ТФ, ранних, частых и тяжелых одышечно-цианотических приступах;
2) при наличии стойкой одышки и тахикардии в покое, не купирующихся консервативной терапией;
3) при нарастании тяжелой дистрофии и анемизации.
Таким больным раннего возраста, а также более старшим детям с тяжелым инфундибулярным стенозом или гипоплазией легочной артерии (диаметр ЛА менее 1/4 диаметра восходящей аорты), показана паллиативная шунтирующая операция по созданию аорто-легочного анастомоза (фактически искусственного артериального протока).
Относительными противопоказаниями к операции являются:
1) острые нарушения мозгового кровообращения;
2) манифестно протекающий инфекционный эндокардит;
3) выраженная сердечная декомпенсация;
4) аноксическая кахексия с нарушением функций всех органов;
5) активный туберкулез легких и др.
У 60—70% больных результаты операций хорошие или значительно улучшается состояние. Смертность в настоящее время не превышает 5— 10% (Подзолков В.П. и др., 1986; Бураковский В.И. и др., 1996; Затикян Е.П., 1996). После операции значительно уменьшаются одышка и цианоз, как правило, исчезают одышечно-цианотические приступы. Дети начинают лучше прибавлять в массе, становятся более активными. Физикально выслушивается типичный систолодиастолический шум аорто-легочного анастомоза, на основании сердца и в межлопаточном пространстве на спине. Позже улучшаются наполнение и выраженность легочного рисунка на рентгенограммах.
Другой паллиативный вариант операции заключается в реконструктивной пластике путей оттока из правого желудочка, без закрытия ДМЖП. К такой коррекции прибегают у больных с гипоплазией легочной артерии при невозможности проведения радикальной операции и малой шунтирующей эффективности паллиативных аорто-легочных анастомозов (Подзолков В.П. и др., 1987).
Радикальная операция включает в себя одновременное устранение стеноза и пластику выводного отдела правого желудочка, закрытие большого дефекта межжелудочковои перегородки с помощью синтетических (тефлон, дакрон) или биологических (ксеноперикард) заплат. Помещение заплаты правее устья аорты, одновременно с закрытием ДМЖП корригирует и декстропозицию аорты. Операция показана как второй этап хирургической коррекции, через 2—3 года после первой, паллиативной, операции, но не позже, чем в 6—7-летнем возрасте.
Первичная радикальная операция показана больным старше 3 лет «бледной» формой ТФ или при умеренной выраженности симптоматики порока. Она проводится успешно лишь в крупных кардиохирургических центрах, однако и там послеоперационная летальность превышает 10—15% (Бураковский В.И. и др., 1996; Asano К. et al., 1985). Операция проводится в режиме искусственного кровообращения в сочетании с регулируемой гипотермией тела.
Оптимальным для радикальной операции считается возраст больного от 4 до 7 лет. Сложность различных анатомических вариантов порока и большой объем реконструктивных и пластических вмешательств увеличивают риск неблагоприятных исходов у детей более раннего возраста. Однако в настоящее время выявляется четкая тенденция к снижению госпитальной летальности после первичной или двухэталной радикальной операции, которая уменьшилась до 5—10%.
В то же время при высоких степенях инфундибулярного стеноза либо при сочетании его с гипоплазией ствола легочной артерии и ее ветвей или с другими сердечными аномалиями летальность может возрастать до 50% (Kirklin J., Barrat-Boyes В., 1986).
Отдаленные результаты после радикальной коррекции порока у 78—95% больных хорошие. У них исчезает цианоз, значительно уменьшаются одышка, полицитемия, с годами проходит деформация ногтей и фаланг пальцев, дети ведут активный образ жизни, приближающийся к активности нетренированных здоровых детей, занимаются лечебной физкультурой, посещают учебные заведения, и около 90% пациентов живут более 20 лет после радикальной операции (Бураковскии В.И. и др., 1996).
Физикально выявляется значительное уменьшение интенсивности, продолжительности и иррадиации систолического шума стеноза, но почти у 90% пациентов он все-таки выслушивается локально во втором-третьем межреберьях слева от грудины. У большинства больных там же может выслушиваться интенсивный протодиастолический шум пульмональной недостаточности, обусловленной травматическим повреждением створок, пульмонального клапана при его пластической коррекции.
Большинство кардиохирургов в настоящее время рекомендуют корригировать порок в грудном возрасте, так как, вопреки распространенному мнению, ТФ значительно ухудшает состояние больных уже в раннем возрасте. Летальность при ранних радикальных операциях не превышает 10%, а отдаленные результаты лучше, чем у пациентов, оперированных в более старшем возрасте, у которых возрастает частота поздних нарушении сердечного ритма и сократимости миокарда (Егорова И.Ф. и др., 1999; Starnes V. et al., 1994). Но проведение операций в таком возрасте возможно лишь в клиниках, где накоплен достаточный опыт.
7. Учебно-методический материал:

Рис. 1. Схематическое изображение различных вариантов расположения ложных хорд левого желудочка сердца (по Т.Ф.Перетолчиной, 1996; Э.В.Земцовскому, 2000).
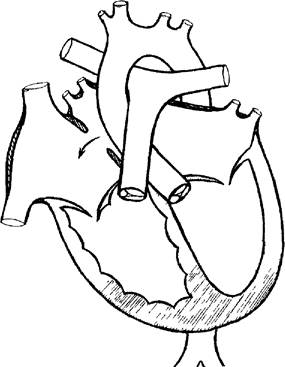
Рис. 2. Схема сердца с вторичным дефектом межпредсердной перегородки (стрелкой указано направление шунта).
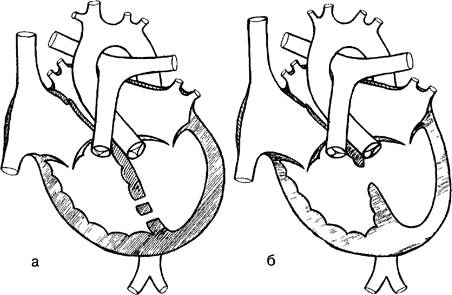
Рис. 3. Схема сердца с дефектом межжелудочковой перегородки. (а — ДМЖП в мышечной части; б — ДМЖП перимембранозный).
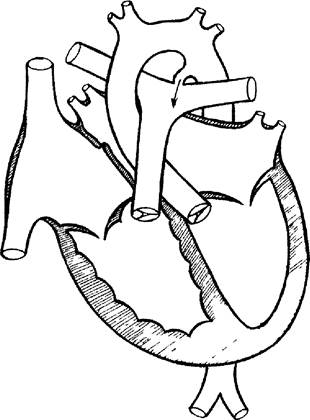
Рис. 4. Схема сердца с открытым артериальным протоком.
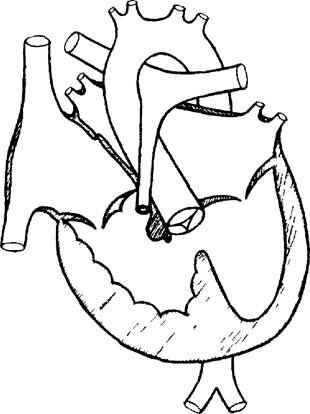
Рис. 5. Схема сердца с тетрадой Фалло.
8. Задания в тестовой форме:
1) к атавистическим аномалиям развития человека относятся:
А) расщелина твердого неба – “волчья пасть”;
Б) ихтиоз;
В) трехкамерное сердце;
Г) макроцефалия;
Д) сохранение двух дуг аорты.
2) нарушение клеточной адгезии является ведущим механизмом в формировании:
А) тазового расположения почки;
Б) spina bifida;
В) “заячьей губы”;
Г) полидактилии;
Д) крипторхизма.
3) антимонголоидный разрез глаз – это:
А) увеличение расстояния между внутренними углами глазных щелей;
Б) опущенные наружные углы глазных щелей;
В) узкая глазная щель;
Г) опущенные внутренние углы глазных щелей;
Д) полулунная складка у внутреннего угла глаза.
4) По этиологическому признаку врожденные пороки различают:
А) наследственные;
Б) дизонтогенетические;
В) экзогенные;
Г) мультифакториальные;
Д) филогенетические.
5) среди врожденных пороков, связанных с аномалией расположения органа, различают:
А) агенезия;
Б) гамартия;
В) гетеротопия;
Г) дистопия;
Д) инверсия.
6) анатомическими признаками тетрады фалло являются:
А) стеноз аорты;
Б) стеноз легочной артерии;
В) смещение аорты вправо;
Г) дефект межпредсердной перегородки;
Д) дефект межжелудочковой перегородки.
7) открытое овальное отверстие является отклонением от нормы в случае, если оно обнаруживается в возрасте:
А) старше 1 года;
Б) до 1 года;
В) старше 5 лет;
Г) в любом возрасте.
8) группа ВПС, для которых характерно наличие аномального сообщения между двумя предсердными камерами, называется:
А) дефект межжелудочковой перегородки (ДМЖП);
Б) коарктация;
В) открытое овальное окно;
Г) болезнь Фалло;
Д) дефект межпредсердной перегородки (ДМПП).
9) в понятие синдрома эйзенменгера входят:
А) субаортальная локализация дефекта межжелудочковой перегородки;
Б) мышечная локализация дефекта межжелудочковой перегородки;
В) дилатация ствола легочной артерии;
Г) снижение давления в малом круге кровообращения;
Д) повышение давления в малом круге кровообращения;
Е) стеноз аорты.
10) Открытый артериальный проток относится к порокам:
А) бледного типа с обеднением малого круга кровообращения;
Б) бледного типа с обогащением малого круга кровообращения;
В) синего типа с обеднением малого круга кровообращения;
Г) синего типа с обогащением малого круга кровообращения.
Эталоны ответов на тесты:
1. А, В, Д
2. Б, В
3. Б
4. А, В, Г
5. В, Г, Д
6. Б, В, Д
7. А
8. Д
9. А, В, Д
10. Б
9. Ситуационные задания:
Задание 1.
Мальчик К., 11 месяцев, поступил в стационар с жалобами на отставание в физическом развитии (масса тела 7 кг), появление одышки и периорального цианоза при физической активности.
Из анамнеза известно, что недостаточная прибавка в массе тела отмечается с 2-х месячного возраста, при кормлении отмечалась быстрая утомляемость вплоть до отказа от груди. При осмотре: кожные покровы с цианотичным оттенком, периферический цианоз, симптом "барабанных палочек" и "часовых стекол". В легких пуэрильное дыхание, хрипов нет, ЧД - 40 в мин. Область сердца не изменена, границы сердца: правая - по правой парастернальной линии, верхняя - II межреберье, левая - по левой средне-ключичной линии. Тоны сердца ритмичные, ЧСС - 140 в мин., вдоль левого края грудины выслушивается систолический шум жесткого тембра, II тон ослаблен во втором межреберье слева. Живот мягкий, безболезненный при пальпации. Печень и селезенка не увеличены. Стул, диурез в норме.
Общий анализ крови: гематокрит - 49%, НЬ 170 г/л, Эр –5,4*1012/л, Ц.п. - 0,91, Лейкоциты – 6,1*109/л, п/я - 3%, с - 26% э - 1%, л - 64%, м - 6%, СОЭ - 2 мм/час.
Задание:
1. Сформулируйте предварительный диагноз.
2. Перечислите дополнительные методы обследования для подтверждения диагноза.
3. С чем связаны повышенные значения уровней гематокрита, гемоглобина и эритроцитов?
Эталоны ответов на ситуационные задания:
Задание 1.
1. Врожденный порок сердца: тетрада Фалло, НК IIб.
2. Эхокардиография, обзорная рентгенография органов грудной клетки, электрокардиография.
3. Повышенные уровни эритроцитов, гемоглобина и гематокрита связаны с гипоксией в крови в результате порока сердца.
10. Литература:
1. Диагностика и лечение врожденных и наследственных заболеваний у детей (путеводитель по клинической генетике) / Ю.И.Барашнев, В.А.Бахарев, П.В.Новиков. – М.: “Триада-Х”, - 2004, - 560с.
2. Медицинская генетика: учеб.-метод. пособие / А. Ф. Бабцева [и др.] // Благовещенск, АГМА, - 2002, - 76 с.
3. Тератология человека / под ред. Г.И. Лазюка, - М., - 1979.
4. Пороки и малые аномалии сердца у детей и подростков / О.А.Мутафьян, - СПб.: Издательский дом СПбМАПО, - 2005, - 480с.
5. Учебник под ред. Баранова А.А. / Детские болезни.- М: ГЭОТАР-Медиа, 2009.- 1008с.
Настраиваем раздачу интернета по Wi-Fi с ноутбука на Windows 8 и Windows 8.1. Настройка точки доступа
В этой статье вы найдете всю необходимую информацию по настройке точки доступа на Windows 8 и Windows 8.1. Мы настроим раздачу интернета с ноутбука, который работает на Windows 8 и подключим к этому ноутбуку по Wi-Fi наш телефон, планшет, другой компьютер, телевизор и т. д. Наш компьютер будет выполнять роль Wi-Fi роутера.
 Я уже очень давно собирался подготовить эту статью, так как это очень популярная тема на данный момент. Статья по настройке точки доступа на Windows 7 собрала очень много просмотров и комментариев. Да и инструкции по раздаче интернета с Android устройств и даже Smart TV, тоже популярные.
Я уже очень давно собирался подготовить эту статью, так как это очень популярная тема на данный момент. Статья по настройке точки доступа на Windows 7 собрала очень много просмотров и комментариев. Да и инструкции по раздаче интернета с Android устройств и даже Smart TV, тоже популярные.
Коротко о том, что мы будем делать, и для чего вообще запускать точку доступа на ноутбуке.
Например, у вас есть ноутбук (возможно персональный компьютер с Wi-Fi адаптером), есть проводной интернет (или интернет через 3G/4G модем), и устройства, которые можно подключать к интернету по Wi-Fi. Современные смартфоны, планшеты, ноутбуки, телевизоры, практически все имеют такую возможность. А у вас интернет по кабелю, или от USB модема.
Мы подключаем интернет к компьютеру на котором есть Wi-Fi и запускаем на нем точку доступа. В этой статье мы будем делать это на примере Windows 8. Вот и все, наш ноутбук получает интернет по кабелю, и раздает по Wi-Fi на нужные нам устройства. Вот и вся схема  . Если вы не хотите тратить деньги на покупку Wi-Fi роутера, или он просто вам не нужен (например, очень редко нужен Wi-Fi), то этот способ, то что вам нужно.
. Если вы не хотите тратить деньги на покупку Wi-Fi роутера, или он просто вам не нужен (например, очень редко нужен Wi-Fi), то этот способ, то что вам нужно.
Настройка раздачи интернета на Windows 8 практически никак не отличается от настройки на Windows 7 (ссылка на инструкцию есть в начале этой статьи). Но, я понимаю, что намного проще делать все по инструкции, которая написана конкретно для операционной системы, которая установлена на вашем ноутбуке. А так как Windows 8 (8.1)активно набирает обороты, то эта статья будет полезной.
Дата добавления: 2015-03-03; просмотров: 1069;