Понятие и задачи операционного бенчмаркинга
5.1. Общее описание многоэлектронной системы.
Формальное применение квантовой механики для описания многоэлектронных систем – атомов и молекул, не представляет трудностей. Для них можно построить гамильтониан и записать соответствующее уравнение Шрёдингера. Однако решение такого уравнения оказывается невозможным. Это связано с тем, что не существует такой системы координат, в которой можно было бы разделить переменные (координаты электронов) в многоэлектронном уравнении. Невозможность такого разделения связана с существованием электростатического взаимодействия между электронами, которым нельзя пренебречь. Поскольку решение уравнения Шрёдингера путём разделения переменных невозможно, были развиты различные подходы к описанию электронного строения атомов и молекул (адиабатическое приближение, одноэлектронное приближение и т.д.). Хотя это чисто математические приёмы, однако именно они позволили сформировать понятийный аппарат современной квантовой химии. В общем случае под многоэлектронной системой понимают систему, состоящую из 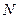 электронов и
электронов и 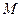 ядер. Если
ядер. Если 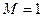 , то рассматриваемая система будет представлять собой многоэлектронный атом. Если же
, то рассматриваемая система будет представлять собой многоэлектронный атом. Если же 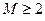 , то система является молекулярной (молекула, радикал, ион). В связи с этим для многоэлектронной системы, при условии что , можно использовать общий термин - молекулярная система. Полная волновая функция молекулярной системы зависит от пространственных (орбитальных) и спиновых координат электронов, а также от пространственных координат ядер:
, то система является молекулярной (молекула, радикал, ион). В связи с этим для многоэлектронной системы, при условии что , можно использовать общий термин - молекулярная система. Полная волновая функция молекулярной системы зависит от пространственных (орбитальных) и спиновых координат электронов, а также от пространственных координат ядер:
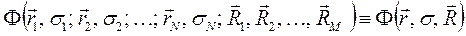
здесь 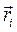 и
и 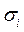 - пространственные и спиновые координаты электронов;
- пространственные и спиновые координаты электронов; 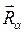 - координаты ядер рассматриваемой многоэлектронной системы. Как и во всех других случаях, гамильтониан молекулярной системы
- координаты ядер рассматриваемой многоэлектронной системы. Как и во всех других случаях, гамильтониан молекулярной системы 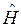 записывается по образцу классического
записывается по образцу классического 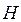 и включает в себя операторы двух типов – кинетической
и включает в себя операторы двух типов – кинетической 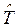 и потенциальной
и потенциальной 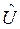 энергии. Как известно, кинетическая энергия – аддитивная величина, она равна сумме кинетической энергии ядер
энергии. Как известно, кинетическая энергия – аддитивная величина, она равна сумме кинетической энергии ядер 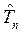 и электронов
и электронов 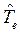 . Потенциальную энергию системы заряженных частиц можно записать как сумму трёх слагаемых: потенциальной энергии притяжения электронов к ядрам,
. Потенциальную энергию системы заряженных частиц можно записать как сумму трёх слагаемых: потенциальной энергии притяжения электронов к ядрам, 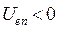 ; потенциальной энергии межэлектронного отталкивания,
; потенциальной энергии межэлектронного отталкивания, 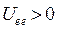 и потенциальной энергии межъядерного отталкивания,
и потенциальной энергии межъядерного отталкивания, 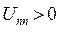 . Тогда оператор Гамильтона для молекулярной системы как совокупность соответствующих операторов, будет иметь следующую структуру:
. Тогда оператор Гамильтона для молекулярной системы как совокупность соответствующих операторов, будет иметь следующую структуру:
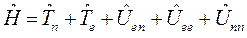
Как известно, масса ядра намного больше массы электронов. В силу этого, период ядерных колебаний на несколько порядков превышает период электронных колебаний. В связи с этим молекулу можно рассматривать состоящей из двух подсистем: медленной (ядерной) и электронной (быстро меняющейся) подсистем. Медленно движущаяся система ядер образует электростатическое поле, в котором с намного большей скоростью движутся электроны. Различие скоростей движения этих подсистем настолько велико, что электроны практически мгновенно реагируют на изменения в положении ядер в пространстве, успевая «подстроиться» к этому изменению. В 1927 г. М. Борн и Р. Оппенгеймер предложили упрощённый подход, в рамках которого электронное движение полностью отделяют от движения ядер. В адиабатическом приближении также рассматривают отдельно электронные и ядерные движения, однако учитывают их слабое взаимодействие. В квантовой механике адиабатическое приближение определяет уравнения, которые описывают движение электронов (точнее говоря, их состояния) при фиксированных значениях ядерных координат. Действительно, если принять массу ядер бесконечно большой, их скорости становятся равными нулю, а это значит, что будет равна нулю и их кинетическая энергия 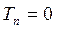 . Таким образом, в адиабатическом приближении движения электронов и ядер считают независимыми, полагая ядра жёстко фиксированными. Тогда волновая функция
. Таким образом, в адиабатическом приближении движения электронов и ядер считают независимыми, полагая ядра жёстко фиксированными. Тогда волновая функция 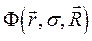 может быть записана в виде произведения электронной
может быть записана в виде произведения электронной 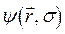 и ядерной (или колебательной)
и ядерной (или колебательной) 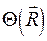 функций:
функций:
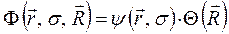
здесь представляет собой ядерную функцию, которая зависит только от координат ядер 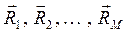 ; а - электронная функция, зависящая от координат электронов
; а - электронная функция, зависящая от координат электронов 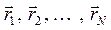 . Необходимо заметить, что координаты ядер входят в электронную функцию как фиксированные параметры, это означает, в свою очередь, что
. Необходимо заметить, что координаты ядер входят в электронную функцию как фиксированные параметры, это означает, в свою очередь, что 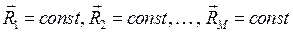 . На основании приведенных выше выкладок, несложно записать уравнение Шрёдингера для молекулярной системы в операторном виде:
. На основании приведенных выше выкладок, несложно записать уравнение Шрёдингера для молекулярной системы в операторном виде:
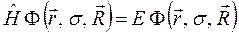
где 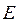 - полная энергия молекулярной системы. В адиабатическом приближении данное уравнение распадается соответственно на два уравнения вида: уравнение движения электронов в поле фиксированных ядер (электронное уравнение) и уравнение движения ядер (ядерное уравнение). В квантовой химии решают электронное уравнение:
- полная энергия молекулярной системы. В адиабатическом приближении данное уравнение распадается соответственно на два уравнения вида: уравнение движения электронов в поле фиксированных ядер (электронное уравнение) и уравнение движения ядер (ядерное уравнение). В квантовой химии решают электронное уравнение:
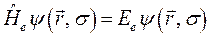
здесь:
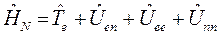
Таким образом, гамильтониан многоэлектронной системы при фиксированном положении ядер можно рассматривать состоящим соответственно из вклада кинетической энергии всех 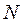 электронов, электростатического притяжения каждого
электронов, электростатического притяжения каждого 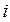 -го электрона ко всем
-го электрона ко всем 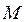 ядрам с зарядами
ядрам с зарядами 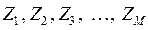 и координатами
и координатами 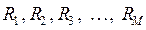 , электростатического отталкивания электронов, а также кулоновское отталкивание ядер. Последнее впрочем не повлияет на решение электронной задачи, но должно учитываться в более точных расчётах, т.е. имеем:
, электростатического отталкивания электронов, а также кулоновское отталкивание ядер. Последнее впрочем не повлияет на решение электронной задачи, но должно учитываться в более точных расчётах, т.е. имеем:
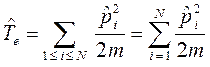
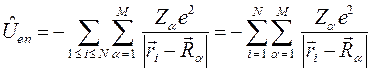
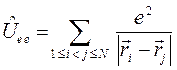
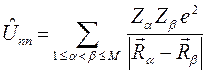
Ядерное (или колебательное) уравнение служит основой задач о колебаниях и вращениях молекул (ИК - спектроскопия, спектры КР и т.д.):
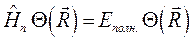
здесь:
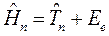
Полная энергия молекулярной системы, очевидно, будет представлять собой сумму электронной энергии, вычисленную при фиксированном расположении ядер в пространстве, и ядерной составляющей :
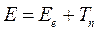
В адиабатическом приближении электронная энергия 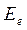 зависит от взаимного расположения ядер, причём эта же энергия как функция координат ядер может быть интерпретирована как потенциальная энергия электронов в поле ядер,
зависит от взаимного расположения ядер, причём эта же энергия как функция координат ядер может быть интерпретирована как потенциальная энергия электронов в поле ядер, 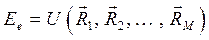 . В системе координат
. В системе координат 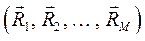 функции
функции 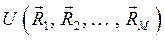 соответствует некоторая поверхность, которую называют поверхностью потенциальной энергии (ППЭ). Приближение Борна-Оппенгеймера и адиабатическое приближение теряют смысл для вырожденных или квазивырожденных электронных состояний. В случае вырождения, электронные уровни смешиваются с колебательными и вращательными уровнями. Такие состояния называют также ровибронными. Для молекулярной системы, состоящей из ядер и электронов, введём обозначения, которых будем придерживаться в последующих выкладках:
соответствует некоторая поверхность, которую называют поверхностью потенциальной энергии (ППЭ). Приближение Борна-Оппенгеймера и адиабатическое приближение теряют смысл для вырожденных или квазивырожденных электронных состояний. В случае вырождения, электронные уровни смешиваются с колебательными и вращательными уровнями. Такие состояния называют также ровибронными. Для молекулярной системы, состоящей из ядер и электронов, введём обозначения, которых будем придерживаться в последующих выкладках: 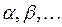 - номера ядер в молекулярной системе;
- номера ядер в молекулярной системе; 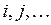 - номера электронов. Тогда одноэлектронный гамильтониан для электрона с номером
- номера электронов. Тогда одноэлектронный гамильтониан для электрона с номером 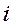 включающий в себя оператор кинетической энергии и оператор потенциальной энергии притяжения электрона ко всем ядрам системы
включающий в себя оператор кинетической энергии и оператор потенциальной энергии притяжения электрона ко всем ядрам системы 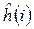 и оператор межэлектронного взаимодействия, двухэлектронный оператор
и оператор межэлектронного взаимодействия, двухэлектронный оператор 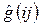 – отталкивания двух электронов
– отталкивания двух электронов 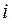 и
и 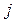 могут быть записаны в виде:
могут быть записаны в виде:
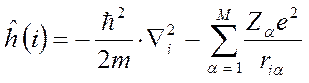
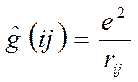
С учётом выражений полученных выше, выражение для электронного гамильтониана молекулярной системы можно представить в виде:
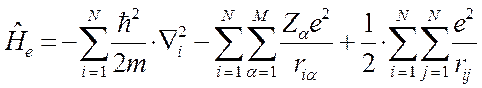
или в более компактной форме:
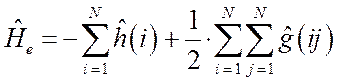
В полученном нами выше выражении символ двойной суммы, в общем случае означает, что суммирование проводится по всем электронам и, следовательно, сумма состоит из слагаемых:
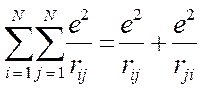
а также учитывая, что:
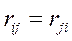
то каждое взаимодействие в такой сумме учтено дважды. При этом правая и левая части соотношения не равны друг другу, т.к. здесь каждое взаимодействие учтено дважды:
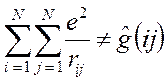
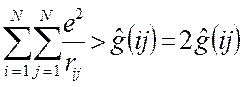
Для того чтобы учесть каждое взаимодействие один раз, перед выражением для потенциальной энергии ставят множитель 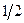 , т.е. имеем:
, т.е. имеем:
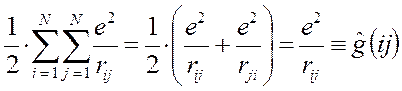
Электронный гамильтониан молекулярной системы всегда содержит слагаемое, отвечающее отталкиванию ядер. Поскольку в адиабатическом приближении расстояния между ядрами строго фиксированы, это слагаемое входит в гамильтониан как параметр и для простоты изложения обычно не учитывается, однако при точных количественных расчётах этот учёт необходим.
5.2. Волновая функция многоэлектронной системы.
Тождественность частиц. Принцип Паули.
В связи с тем, что мы имеем дело с многоэлектронными системами (атомами, молекулами, комплексами и т.д.), возникает вопрос о структуре и свойствах многоэлектронной волновой функции. В квантовой механике он решается на основе двух важнейших принципов – принципа тождественности частиц и принципа Паули. В системе одинаковых (тождественных) частиц реализуются только такие состояния, при которых наблюдаемые физические характеристики системы не изменяются при обмене частиц координатами. В справедливости данного принципа не трудно убедиться. Действительно, пусть у нас имеется некоторая двухэлектронная система. Полную волновую функцию такой системы обозначим, как 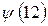 и будем называть её двухэлектронной волновой функцией. Здесь символ
и будем называть её двухэлектронной волновой функцией. Здесь символ 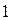 соответствует совокупности всех координат (пространственных и спиновых) первой частицы, а символ
соответствует совокупности всех координат (пространственных и спиновых) первой частицы, а символ 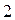 - совокупности всех координат второй частицы. В силу принципа тождественности перестановка частиц местами не должна сказаться на значениях наблюдаемых физических величин. Математически такая перестановка соответствует обмену частиц местами, т.е. переходу от функции к функции
- совокупности всех координат второй частицы. В силу принципа тождественности перестановка частиц местами не должна сказаться на значениях наблюдаемых физических величин. Математически такая перестановка соответствует обмену частиц местами, т.е. переходу от функции к функции 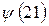 . Если эти функции являются собственными функциями гамильтониана, то они должны соответствовать одному и тому же собственному значению полной энергии системы
. Если эти функции являются собственными функциями гамильтониана, то они должны соответствовать одному и тому же собственному значению полной энергии системы 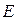 :
:
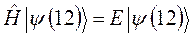
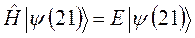
При этом средние значения для оператора 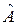 , вычисленные с функциями и , также должны совпадать:
, вычисленные с функциями и , также должны совпадать:
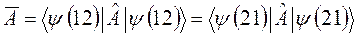
Симметричные волновые функции при перестановке координат двух частиц местами не меняют знак, а антисимметричные – меняют. Очевидно, что свойствами:
обладают как симметричные, так и антисимметричные волновые функции. Это означает, в свою очередь, что:
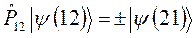
здесь 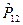 - оператор перестановки координат двух электронов. Симметричным волновым функциям соответствует знак плюс, а антисимметричным – знак минус. Из свойств симметрии волновых функций следует принцип Паули: «Волновая функция электронной системы должна быть антисимметричной относительно перестановки любой пары частиц местами». Это в свою очередь означает, что в квантовом состоянии, определяемом с помощью четырёх квантовых чисел
- оператор перестановки координат двух электронов. Симметричным волновым функциям соответствует знак плюс, а антисимметричным – знак минус. Из свойств симметрии волновых функций следует принцип Паули: «Волновая функция электронной системы должна быть антисимметричной относительно перестановки любой пары частиц местами». Это в свою очередь означает, что в квантовом состоянии, определяемом с помощью четырёх квантовых чисел  ,
,  ,
, 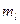 и
и 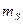 , может находиться не более одного электрона. Данный принцип следует из требования антисимметрии электронной волновой функции. Следствием принципа Паули является так называемый запрет Паули, согласно которому, на одной атомной орбитали могут находиться не более чем два электрона, с противоположно направленными спинами (т.е. имеющими различные значения спиновых квантовых чисел). В соответствии с принципом Паули для двухэлектронной системы, выражение вида:
, может находиться не более одного электрона. Данный принцип следует из требования антисимметрии электронной волновой функции. Следствием принципа Паули является так называемый запрет Паули, согласно которому, на одной атомной орбитали могут находиться не более чем два электрона, с противоположно направленными спинами (т.е. имеющими различные значения спиновых квантовых чисел). В соответствии с принципом Паули для двухэлектронной системы, выражение вида:
должно быть записано в виде:
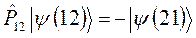
Необходимо отметить, что принцип Паули является важнейшим принципом квантовой механики. Он не является следствием основных её законов, а является утверждением, обобщающим экспериментальные факты. Это означает, что электронных систем, которые были бы симметричны по отношению к перестановке электронов местами, просто не существует. Для того чтобы принцип Паули соблюдался, частицы должны обладать полуцелым спином. К таким частицам, обладающим полуцелым спином и подчиняющимся принципу Паули – относятся фермионы, для которых справедливо распределение Ферми-Дирака. Фермионы находятся только в антисимметричных состояниях. К ним относятся электроны, а также ядра с нечётным числом частиц. Другой класс тождественных частиц – бозоны. К ним относятся  – частицы, ядра с чётным числом частиц, атом водорода, атом гелия и т.д. Такие частицы обладают целочисленным (в том числе и нулевым) спином, а их волновые функции являются симметричными относительно перестановки координат двух частиц. Для бозонов справедлива другая статистика – Бозе-Эйнштейна, описываемая функцией. Поскольку согласно принципу Паули в каждом квантовом состоянии может находиться не более одного электрона, и если имеется система, состоящая из двух электронов, то возникает вопрос относительно вида волновой функции
– частицы, ядра с чётным числом частиц, атом водорода, атом гелия и т.д. Такие частицы обладают целочисленным (в том числе и нулевым) спином, а их волновые функции являются симметричными относительно перестановки координат двух частиц. Для бозонов справедлива другая статистика – Бозе-Эйнштейна, описываемая функцией. Поскольку согласно принципу Паули в каждом квантовом состоянии может находиться не более одного электрона, и если имеется система, состоящая из двух электронов, то возникает вопрос относительно вида волновой функции 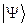 , описывающей её состояние. Предположим, что состояние каждого из электронов задаётся своей волновой функцией
, описывающей её состояние. Предположим, что состояние каждого из электронов задаётся своей волновой функцией 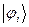 , зависящей от пространственных
, зависящей от пространственных 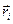 и спиновых
и спиновых 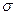 координат, т.е.
координат, т.е.
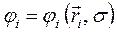
Обозначим всю совокупность пространственных и спиновых координат каждого из электронов соответственно символами 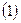 и
и 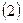 . На основании теоремы о невзаимодействии, согласно которой «если система состоит из двух или нескольких невзаимодействующих подсистем, то её полная волновая функция
. На основании теоремы о невзаимодействии, согласно которой «если система состоит из двух или нескольких невзаимодействующих подсистем, то её полная волновая функция 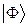 равна произведению волновых функций отдельных подсистем
равна произведению волновых функций отдельных подсистем 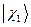 ,
, 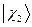 ,
, 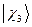 , …,
, …, 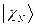 , а энергия – сумме энергий подсистем
, а энергия – сумме энергий подсистем 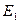 ,
, 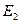 ,
, 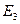 , …,
, …, 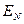 »:
»:
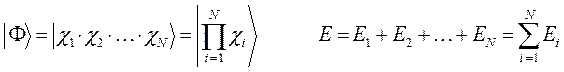
или для нашего случая:
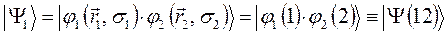
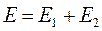
Поскольку все электроны тождественны и неразличимы, то обмен двух электронов местами не повлияет на энергию системы. Следовательно, в том же энергетическом состоянии рассматриваемую систему, состоящую из двух электронов можно описать функцией, образованной из 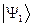 перестановкой координат электронов. Для этого подействуем на исходную функцию оператором перестановки координат соответствующих электронов , получим:
перестановкой координат электронов. Для этого подействуем на исходную функцию оператором перестановки координат соответствующих электронов , получим:
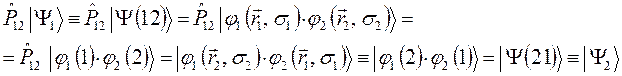
Итак, одному и тому же энергетическому состоянию отвечают две функции и 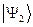 , различающиеся между собой перестановкой координат. Каждая из них представляет собой частные, линейно независимые решения волнового уравнения Шрёдингера. Для того чтобы электронная волновая функция удовлетворяла принципу Паули, необходимо выполнение условия, согласно которому:
, различающиеся между собой перестановкой координат. Каждая из них представляет собой частные, линейно независимые решения волнового уравнения Шрёдингера. Для того чтобы электронная волновая функция удовлетворяла принципу Паули, необходимо выполнение условия, согласно которому:
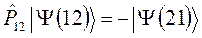
тогда соответственно из двух общих решений:
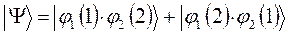
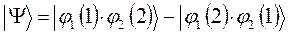
являющихся линейной комбинацией соответствующих частных решений:
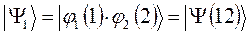
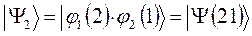
выбираем единственно возможное, построенное в соответствии с принципом антисимметрии:
Решение со знаком «+» является симметричным по отношению к перестановке координат электронов ( при этом не меняет знака). Второе решение со знаком «-» называют антисимметричным (при перестановке координат функция меняет свой знак на противоположный), в чём достаточно легко убедиться. Для этого переставим координаты в соответствующих выражениях для :
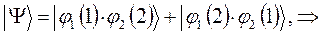
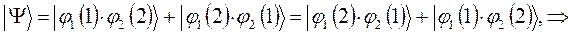
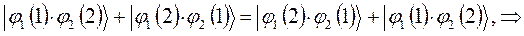
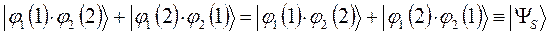
как это очевидно, волновая функция характеризующая состояние электронной системы не изменила свой знак. Такое решение, очевидно, будет являться симметричным относительно перестановки координат. Обозначим данное симметричное состояние 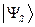 . Аналогично проведём перестановку координат для решения вида:
. Аналогично проведём перестановку координат для решения вида:
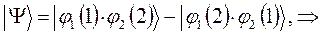
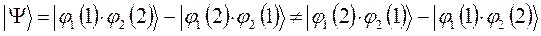
в данном случае приходим к неравенству вида:
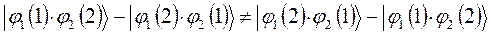
Очевидно, для того чтобы в ходе перестановки координат правая часть данного выражения была равна левой его части, необходимо полученное в ходе перестановки координат выражение заключить в скобки (сгруппировать) и полученный результат умножить на 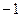 . Выполнив над полученным тождествли соответствующие операции – сгруппировав соответствующие координаты, заключая последние в скобки, умножив полученный результат на , будем иметь:
. Выполнив над полученным тождествли соответствующие операции – сгруппировав соответствующие координаты, заключая последние в скобки, умножив полученный результат на , будем иметь:
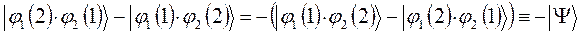
и таким образом:
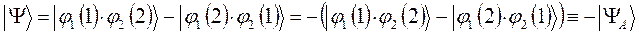
здесь симметричное ( 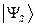 ) и ассиметричное (
) и ассиметричное ( 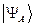 ) решения. Поскольку во втором случае волновая функция меняет свой знак на противоположный, она является антисимметричной относительно операции перестановки координат . Какое из двух решений или отвечает реальной системе? Принцип Паули говорит, что системы, составленные из электронов, описываются исключительно антисимметричными полными волновыми функциями – частица может находиться в каком-либо одном из квантовых состояний. Нахождение двух частиц в одном и том же квантовом состоянии, очевидно, противоречит принципу Паули. Подтверждением справедливости данного принципа служит требование ортогональности волновых функций, согласно которому нахождение двух частиц в одинаковых квантовых состояниях равно нулю. Таким образом, с учётом принципа Паули, следует пользоваться выражением вида:
) решения. Поскольку во втором случае волновая функция меняет свой знак на противоположный, она является антисимметричной относительно операции перестановки координат . Какое из двух решений или отвечает реальной системе? Принцип Паули говорит, что системы, составленные из электронов, описываются исключительно антисимметричными полными волновыми функциями – частица может находиться в каком-либо одном из квантовых состояний. Нахождение двух частиц в одном и том же квантовом состоянии, очевидно, противоречит принципу Паули. Подтверждением справедливости данного принципа служит требование ортогональности волновых функций, согласно которому нахождение двух частиц в одинаковых квантовых состояниях равно нулю. Таким образом, с учётом принципа Паули, следует пользоваться выражением вида:
которое может быть записано в виде детерминанта второго порядка:
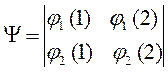
Обобщим теперь задачу построения антисимметричных волновых функций на случай системы из невзаимодействующих электронов. Для этого запишем начальную многоэлектронную функцию в виде произведения одноэлектронных функций:
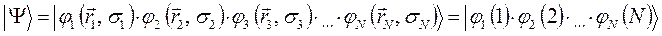
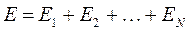
Тогда детерминант, формирующий антисимметричную - электронную волновую функцию, следует записать в виде:
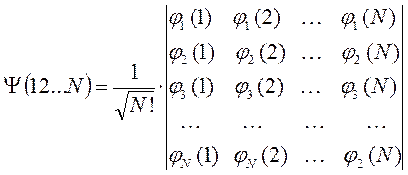
здесь нормировочный множитель 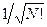 - числовой коэффициент, гарантирующий нормировку многоэлектронной волновой функции
- числовой коэффициент, гарантирующий нормировку многоэлектронной волновой функции 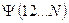 на единицу:
на единицу:
Дж. Слейтер показал, что единственно возможно формой построения полностью антисимметричной волновой функцией - электронной системы из волновых функций отдельных независимых электронов является детерминант - го порядка, который называют детерминантом (определителем) Слейтера, а волновую функцию, записанную в виде детерминанта Слейтера, называют детерминантной. Исходя из свойств определителей, можно показать справедливость принципа Паули, а также нормированность и ортогональность комбинируемых волновых функций. Предположим, что электроны в какой-то момент времени могут оказаться в одном и том же квантовом состоянии, т.е. что 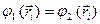 , где
, где 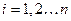 . В теории матриц строго доказывается, что если две строки детерминанта
. В теории матриц строго доказывается, что если две строки детерминанта 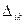 оказываются одинаковыми, то в этом случае детерминант
оказываются одинаковыми, то в этом случае детерминант 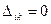 . Полная волновая функция всегда отлична от нуля, т.к. в случае
. Полная волновая функция всегда отлична от нуля, т.к. в случае 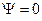 вероятность найти электрон в элементе объёма
вероятность найти электрон в элементе объёма 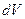 также равна нулю. Поскольку это противоречит требованию нормировки, согласно которому вероятность нахождения электрона в элементе объёма равна единице, поэтому детерминант не может быть равен нулю:
также равна нулю. Поскольку это противоречит требованию нормировки, согласно которому вероятность нахождения электрона в элементе объёма равна единице, поэтому детерминант не может быть равен нулю:
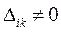
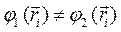
Так, если мы имеем систему невзаимодействующих электронов, то полную энергию такой электронной системы можно представить как сумму энергий отдельных частиц. Действительно, при отсутствии взаимодействия электронов в таких системах, двухэлектронные компоненты гамильтониана 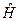 будут равны нулю, т.е.
будут равны нулю, т.е. 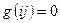 и гамильтониан, таким образом, может быть записан в виде суммы отдельных одноэлектронных операторов
и гамильтониан, таким образом, может быть записан в виде суммы отдельных одноэлектронных операторов 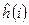 . На основании теоремы о невзаимодействии, согласно которой «если система состоит из двух или нескольких невзаимодействующих подсистем, то её полная волновая функция равна произведению волновых функций отдельных подсистем ,
. На основании теоремы о невзаимодействии, согласно которой «если система состоит из двух или нескольких невзаимодействующих подсистем, то её полная волновая функция равна произведению волновых функций отдельных подсистем , 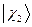 ,
, 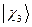 , …,
, …, 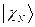 , а энергия – сумме энергий подсистем ,
, а энергия – сумме энергий подсистем , 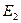 , , …, ».
, , …, ».
имеем соответственно:
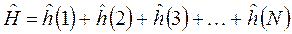
тогда - электронное уравнение Шрёдингера:
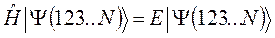
распадается на - одноэлектронных уравнений вида:
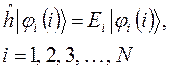
Полная энергия такой системы, при отсутствии межэлектронного взаимодействия, может быть записана в виде суммы:
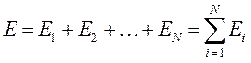
В силу тождественности электронов операторы одинаковы для разных 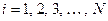 и таким образом, задача о движении невзаимодействующих частиц сводится к задаче о движении одной частицы, которая может быть решена точно. Формально адиабатическое приближение также является следствием независимости электронных и ядерных движений. В отличие от рассмотренных ранее идеализированных моделей, в реальных многоэлектронных системах (атомах, молекулах, ионах, комплексах и т.д.) существует значительное межэлектронное отталкивание, такие системы ещё называют сильно коррелированными. Однако и в этом случае используется модель детерминантной многоэлектронной функции, построенной на одноэлектронных функциях:
и таким образом, задача о движении невзаимодействующих частиц сводится к задаче о движении одной частицы, которая может быть решена точно. Формально адиабатическое приближение также является следствием независимости электронных и ядерных движений. В отличие от рассмотренных ранее идеализированных моделей, в реальных многоэлектронных системах (атомах, молекулах, ионах, комплексах и т.д.) существует значительное межэлектронное отталкивание, такие системы ещё называют сильно коррелированными. Однако и в этом случае используется модель детерминантной многоэлектронной функции, построенной на одноэлектронных функциях:
Действительно, каждому электрону в многоэлектронной системе можно приписать свою индивидуальную волновую функцию (орбиталь) 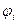 . Это в свою очередь означает, что на каждый отдельный электрон действует единый усреднённый потенциал, создаваемый остальными электронами и ядрами. При этом полная волновая функция системы может быть представлена в виде произведения одноэлектронных функций, которые называют атомными орбиталями в случае атома и молекулярными орбиталями в случае молекулы. Такое приближение к точной многоэлектронной функции называют одноэлектронным (орбитальным) приближением или моделью независимых электронов. Итак, как это было показано уже выше, выражение для электронного гамильтониана молекулярной системы имеет вид:
. Это в свою очередь означает, что на каждый отдельный электрон действует единый усреднённый потенциал, создаваемый остальными электронами и ядрами. При этом полная волновая функция системы может быть представлена в виде произведения одноэлектронных функций, которые называют атомными орбиталями в случае атома и молекулярными орбиталями в случае молекулы. Такое приближение к точной многоэлектронной функции называют одноэлектронным (орбитальным) приближением или моделью независимых электронов. Итак, как это было показано уже выше, выражение для электронного гамильтониана молекулярной системы имеет вид:
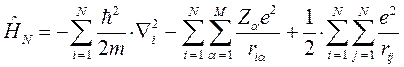
Электронный гамильтониан, очевидно, будет действовать только на пространственные функции и не будет зависеть от спиновых координат. Операторы квадрата спинового углового момента 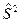 и его проекции
и его проекции 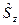 , напротив, действует только на спиновую переменную . В связи с этим, одноэлектронную волновую функцию
, напротив, действует только на спиновую переменную . В связи с этим, одноэлектронную волновую функцию 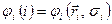 в хорошем приближении можно представить в виде произведения пространственной
в хорошем приближении можно представить в виде произведения пространственной 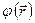 и спиновой
и спиновой 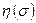 волновых функций:
волновых функций:
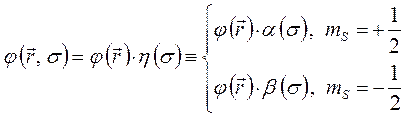
здесь и 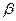 соответствуют двум возможным проекциям спинового магнитного момента на ось
соответствуют двум возможным проекциям спинового магнитного момента на ось 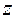 .
.

при этом:

Такую одноэлектронную волновую функцию называют спин-орбиталью. Спин-орбиталь описывает пространственные и спиновые состояния одного электрона. Если же речь идёт о двух электронах, то в силу принципа Паули их полная волновая функция должна быть антисимметрична. На языке спин-орбиталей это означает, что если пространственные волновые функции двух электронов совпадают, то спиновые компоненты должны отличаться:
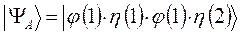
в противном случае мы будем иметь дело с простейшей симметричной двухэлектронной функцией, которую невозможно антисимметризовать, поскольку соответствующий ей детерминант Слейтера будет равен нулю. Действительно, представляя волновую функцию как некоторый вектор состояние вида:
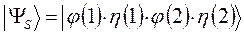
имеем соответственно:
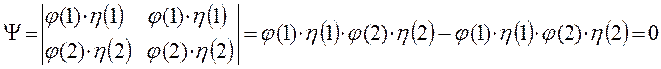
Следовательно, два электрона могут описываться одной пространственной волновой функцией, но при этом их спиновые состояния должны отличаться. И наоборот, если спиновые состояния двух электронов одинаковы, то должны отличаться их пространственные состояния. Это утверждение является следствием принципа Паули для модели независимых электронов. В случае многоэлектронного атома каждая спин-орбиталь характеризуется четырьмя квантовыми числами: 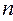 ,
, 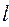 ,
, 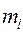 и
и 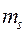 . Причём пространственная волновая функция зависит от трёх квантовых чисел , и , а спиновая – от одного . Учитывая это обстоятельство, принцип Паули можно сформулировать следующим образом: «Не может быть двух электронов с одинаковым набором четырёх квантовых чисел». Два электрона, занимающие одно пространственное состояние и отличающиеся только спиновыми функциями, называют спаренными, а их спин-орбитали записывают в виде:
. Причём пространственная волновая функция зависит от трёх квантовых чисел , и , а спиновая – от одного . Учитывая это обстоятельство, принцип Паули можно сформулировать следующим образом: «Не может быть двух электронов с одинаковым набором четырёх квантовых чисел». Два электрона, занимающие одно пространственное состояние и отличающиеся только спиновыми функциями, называют спаренными, а их спин-орбитали записывают в виде:

Система, состоящая только из спаренных электронов, называется системой с замкнутой оболочкой ( 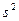 ,
, 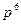 ,
, 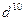 ,
, 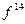 и т.д.). Такая система содержит чётное число электронов, и она может быть описана одной детерминантной волновой функцией. В системах с открытой оболочкой (такие оболочки могут быть как чётно-электронными, так и нечётно-электронными) имеются пространственные состояния (орбитали), которые заняты одним электроном. При этом волновая функция таких систем может быть представлена линейной комбинацией двух или большего числа детерминантов Слейтера. На примере атомов нескольких химических элементов с открытыми и закрытыми оболочками, рассмотрим алгоритм построения такого рода детерминантных функций. Из соображений удобства изложения материала, рассмотрим каким образом можно построить детерминант Слейтера для многоэлектронных систем с замкнутыми оболочками. Как уже говорилось выше, к таким атомам относят системы, состоящие только из спаренных электронов. Такие системы содержат чётное число электронов и могут быть описаны только одной детерминантной волновой функцией. Рассмотрим метод построения такой функции на примере атома бериллия. Построение детерминанта Слейтера для заданной электронной конфигурации атома следует начинать с перечисления всех спин-орбиталей. Так, для основного состояния атома бериллия Be с электронной конфигурацией
и т.д.). Такая система содержит чётное число электронов, и она может быть описана одной детерминантной волновой функцией. В системах с открытой оболочкой (такие оболочки могут быть как чётно-электронными, так и нечётно-электронными) имеются пространственные состояния (орбитали), которые заняты одним электроном. При этом волновая функция таких систем может быть представлена линейной комбинацией двух или большего числа детерминантов Слейтера. На примере атомов нескольких химических элементов с открытыми и закрытыми оболочками, рассмотрим алгоритм построения такого рода детерминантных функций. Из соображений удобства изложения материала, рассмотрим каким образом можно построить детерминант Слейтера для многоэлектронных систем с замкнутыми оболочками. Как уже говорилось выше, к таким атомам относят системы, состоящие только из спаренных электронов. Такие системы содержат чётное число электронов и могут быть описаны только одной детерминантной волновой функцией. Рассмотрим метод построения такой функции на примере атома бериллия. Построение детерминанта Слейтера для заданной электронной конфигурации атома следует начинать с перечисления всех спин-орбиталей. Так, для основного состояния атома бериллия Be с электронной конфигурацией 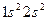 , возможен следующий набор спин-орбиталей:
, возможен следующий набор спин-орбиталей:
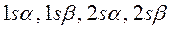
Затем следует записать первую строку детерминанта, указав в круглых скобках, что спин-орбитали относятся к первому электрону. Вторая и последующие строки детерминанта будут отличны друг от друга только номером электрона. В нашем случае имеем детерминант 4-го порядка. Таким образом, порядок детерминанта определяется числом электронов в рассматриваемой многоэлектронной системе. Итак, для атома бериллия имеем соответственно детерминант 4-го порядка:
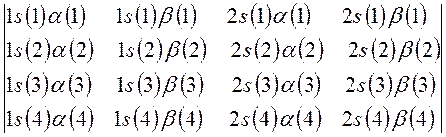
Далее перед детерминантом записывают нормировочный множитель 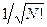 - числовой коэффициент, гарантирующий нормировку многоэлектронной волновой функции
- числовой коэффициент, гарантирующий нормировку многоэлектронной волновой функции 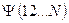 на единицу:
на единицу:
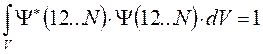
С учётом изложенных выше соображений, несложно записать детерминантную волновую функцию для атома бериллия – многоэлектронной системы с замкнутой оболочкой 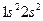 , в виде:
, в виде:
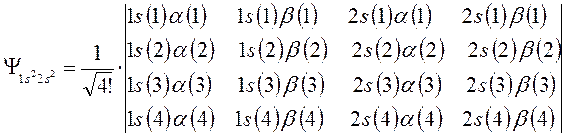
Рассмотрим теперь метод построения детерминантной волновой функции для систем с открытой оболочкой на примере атома лития Li, имеющего электронную конфигурацию 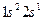 . Напомним, что к системам с открытой оболочкой (такие оболочки могут быть как чётно-электронными, так и нечётно-электронными) относят пространственные состояния (орбитали), которые заняты одним (неспаренным) электроном. При этом волновая функция таких систем может быть представлена линейной комбинацией двух или большего числа детерминантов Слейтера. Как и в предыдущем случае, построение детерминанта Слейтера для заданной электронной конфигурации атома следует начинать с перечисления всех спин-орбиталей. Так, для основного состояния атома лития Li с электронной конфигурацией , возможны два набора спин-орбиталей:
. Напомним, что к системам с открытой оболочкой (такие оболочки могут быть как чётно-электронными, так и нечётно-электронными) относят пространственные состояния (орбитали), которые заняты одним (неспаренным) электроном. При этом волновая функция таких систем может быть представлена линейной комбинацией двух или большего числа детерминантов Слейтера. Как и в предыдущем случае, построение детерминанта Слейтера для заданной электронной конфигурации атома следует начинать с перечисления всех спин-орбиталей. Так, для основного состояния атома лития Li с электронной конфигурацией , возможны два набора спин-орбиталей:
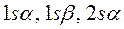
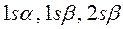
такой учёт становится необходимым в связи с тем, что у атома лития внешний электрон не спарен, по этой причине следует учитывать и 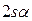 , и
, и 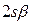 спин-орбитали. После перечисления всех возможных наборов спин-орбиталей, следует записать первую строку детерминанта, указав в круглых скобках, что перечисленные выше спин-орбитали относятся к первому электрону. Все последующие строки будут отличаться от первой, только номером электрона. Так, для каждого из возможных наборов спин-орбиталей:
спин-орбитали. После перечисления всех возможных наборов спин-орбиталей, следует записать первую строку детерминанта, указав в круглых скобках, что перечисленные выше спин-орбитали относятся к первому электрону. Все последующие строки будут отличаться от первой, только номером электрона. Так, для каждого из возможных наборов спин-орбиталей:

будем иметь соответственно:
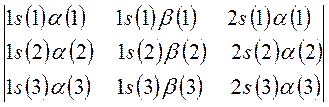
а также:
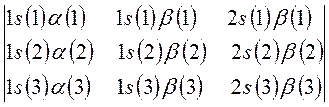
Как и в предыдущем случае, перед детерминантом записывают нормировочный множитель - числовой коэффициент, гарантирующий нормировку многоэлектронной волновой функции на единицу:
где 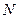 - число электронов в данной многоэлектронной системе. Поскольку рассматриваемая многоэлектронная система является системой с открытой оболочкой, то общая волновая функция такой многоэлектронной системы будет представлять собой линейную комбинацию соответствующего числа детерминантов. При этом число комбинируемых детерминантов можно оценить соотношением вида
- число электронов в данной многоэлектронной системе. Поскольку рассматриваемая многоэлектронная система является системой с открытой оболочкой, то общая волновая функция такой многоэлектронной системы будет представлять собой линейную комбинацию соответствующего числа детерминантов. При этом число комбинируемых детерминантов можно оценить соотношением вида 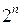 , где
, где 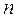 - число комбинируемых детерминантов, равное числу неспаренных электронов (как для чётно-электронных, так и нечётно-электронных систем с открытой оболочкой). Тогда соответственно для атома лития, детерминантная волновая функция, с учётом нормировочного множителя будет иметь вид:
- число комбинируемых детерминантов, равное числу неспаренных электронов (как для чётно-электронных, так и нечётно-электронных систем с открытой оболочкой). Тогда соответственно для атома лития, детерминантная волновая функция, с учётом нормировочного множителя будет иметь вид:
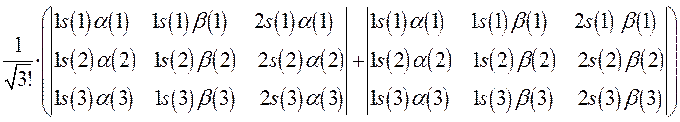
Поскольку каждая из компонент линейной комбинации нормирована на единицу, а также учитывая, что и общая волновая функция, представляющая собой суперпозицию двух детерминантов также должна быть нормирована на единицу, то появляется общий нормировочный множитель 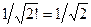 и волновая функция электронной конфигурации приобретает вид:
и волновая функция электронной конфигурации приобретает вид:
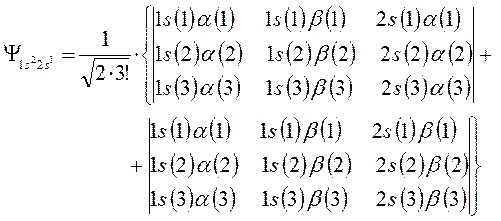
Таким образом, полная (антисимметричная) волновая функция строится как произведение пространственной 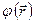 и спиновой
и спиновой 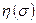 функций. При этом общая антисимметрия волновой функции системы может быть реализована в двух случаях:
функций. При этом общая антисимметрия волновой функции системы может быть реализована в двух случаях:
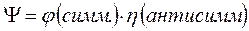
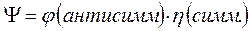
Симметричные спиновые компоненты 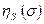 волновой функции
волновой функции 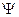 (здесь соответствующие пространственные компоненты
(здесь соответствующие пространственные компоненты 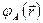 антисимметричны), соответствуют триплетному состоянию (нечётное число электронов):
антисимметричны), соответствуют триплетному состоянию (нечётное число электронов):
Антисимметричные спиновые компоненты волновой функции (здесь соответствующие пространственные компоненты симметричны), соответствуют синглетному состоянию (чётное число электронов):
Итак, как говорилось уже выше, вопрос о структуре и свойствах многоэлектронной волновой функции в рамках адиабатического приближения, может быть решён на основании двух важнейших принципов – принципа тождественности частиц и принципа Паули. Согласно первому из них, в системе одинаковых (тождественных) частиц реализуются только такие состояния, при которых наблюдаемые физические характеристики системы не изменяются при обмене частиц координатами. Согласно принципа Паули, в одном и том же атоме не может быть двух электронов, обладающих одинаковым набором четырёх квантовых чисел , 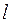 ,
, 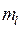 и
и 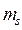 . Исходя из данного принципа, называемого также принципом антисимметрии следует, что волновая функция электронной системы должна быть антисимметричной по отношению к перестановке любой пары частиц местами. Причём общая волновая функция должна учитывать как пространственные, так и спиновые координаты электрона и должна удовлетворять условию антисимметрии, которое аналитически может быть задано детерминантом Слейтера. Так, при рассмотрении электронной конфигурации атома, т.е. распределения в нём электронов по различным квантовым состояниям, исходят из двух фундаментальных принципов – принципа наименьшей энергии и принципа Паули. Согласно принципу наименьшей энергии электроны, присоединяемые к атому, занимают в нём свободные уровни с наименьшей энергией, отвечающие наибольшей их связи с ядром. Принцип наименьшей энергии нашёл своё выражение в так называемых эвристических правилах Клечковского, согласно которым: при переходе от одного элемента к другому электроны размещаются последовательно на атомных орбиталях, расположенных в порядке увеличения суммы главного и орбитального квантовых чисел
. Исходя из данного принципа, называемого также принципом антисимметрии следует, что волновая функция электронной системы должна быть антисимметричной по отношению к перестановке любой пары частиц местами. Причём общая волновая функция должна учитывать как пространственные, так и спиновые координаты электрона и должна удовлетворять условию антисимметрии, которое аналитически может быть задано детерминантом Слейтера. Так, при рассмотрении электронной конфигурации атома, т.е. распределения в нём электронов по различным квантовым состояниям, исходят из двух фундаментальных принципов – принципа наименьшей энергии и принципа Паули. Согласно принципу наименьшей энергии электроны, присоединяемые к атому, занимают в нём свободные уровни с наименьшей энергией, отвечающие наибольшей их связи с ядром. Принцип наименьшей энергии нашёл своё выражение в так называемых эвристических правилах Клечковского, согласно которым: при переходе от одного элемента к другому электроны размещаются последовательно на атомных орбиталях, расположенных в порядке увеличения суммы главного и орбитального квантовых чисел 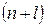 . В случае получения одинаковых значений суммы , в первую очередь заполняются орбитали с меньшим значением главного квантового числа
. В случае получения одинаковых значений суммы , в первую очередь заполняются орбитали с меньшим значением главного квантового числа 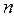 . Порядок расположения атомных орбиталей по энергии определяется последовательностью энергетических уровней:
. Порядок расположения атомных орбиталей по энергии определяется последовательностью энергетических уровней:
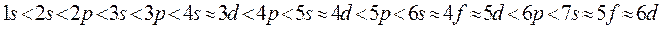
Действительно, как известно, гамильтониан многоэлектронной системы включает в себя члены, учитывающие межэлектронное отталкивание – это так называемые двухэлектронные операторы 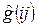 . Это значительно усложняет картину. Так, если энергия электрона в атоме водорода зависит от главного квантового числа , то энергия энергетических уровней в многоэлектронном атоме определяется значениями двух квантовых чисел: главного и орбитального
. Это значительно усложняет картину. Так, если энергия электрона в атоме водорода зависит от главного квантового числа , то энергия энергетических уровней в многоэлектронном атоме определяется значениями двух квантовых чисел: главного и орбитального 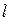 . В связи с этим говорят о расщеплении энергетические уровней с одинаковым значением главного квантового числа , в связи с межэлектронным отталкиванием. Уровни энергии электронов с одинаковым значением главного квантового числа называют электронным слоем (или энергетическим уровнем), а орбитали с одинаковым значением квантового числа - электронной оболочкой (или энергетическим подуровнем). В общем случае при заданном значении главного квантового числа , энергия электронов увеличивается с ростом орбитального квантового числа . Это происходит потому, что с ростом , всё меньшая часть волновой функции сосредоточена в области близкой к ядру, так что усреднённый экранирующий ядро заряд уменьшается. Согласно же принципу Паули волновая функция электронной системы должна быть антисимметричной по отношению к перестановке любой пары частиц местами. Из данного принципа антисимметрии следует важный вывод о том, что в атоме не может быть двух электронов, имеющих одинаковый набор всех четырёх квантовых чисел. Следствием принципа Паули является так называемый запрет Паули, согласно которому на одной атомной орбитали могут находиться не более чем два электрона, имеющие различные значения спиновых квантовых чисел . Данная формулировка опять-таки следует из требования антисимметрии электронной волновой функции.
. В связи с этим говорят о расщеплении энергетические уровней с одинаковым значением главного квантового числа , в связи с межэлектронным отталкиванием. Уровни энергии электронов с одинаковым значением главного квантового числа называют электронным слоем (или энергетическим уровнем), а орбитали с одинаковым значением квантового числа - электронной оболочкой (или энергетическим подуровнем). В общем случае при заданном значении главного квантового числа , энергия электронов увеличивается с ростом орбитального квантового числа . Это происходит потому, что с ростом , всё меньшая часть волновой функции сосредоточена в области близкой к ядру, так что усреднённый экранирующий ядро заряд уменьшается. Согласно же принципу Паули волновая функция электронной системы должна быть антисимметричной по отношению к перестановке любой пары частиц местами. Из данного принципа антисимметрии следует важный вывод о том, что в атоме не может быть двух электронов, имеющих одинаковый набор всех четырёх квантовых чисел. Следствием принципа Паули является так называемый запрет Паули, согласно которому на одной атомной орбитали могут находиться не более чем два электрона, имеющие различные значения спиновых квантовых чисел . Данная формулировка опять-таки следует из требования антисимметрии электронной волновой функции.
5.3. Атомные термы. Векторная модель строения атома.
Оба данных условия (принцип наименьшей энергии и принцип Паули), определяют правила построения электронных конфигураций атома. Однако руководствуясь только лишь данными принципами, не получается в полной мере охарактеризовать электронную конфигурацию атомов многоэлектронных систем. Действительно, для однозначного описания состояния атомов, не достаточно указать его электронную конфигурацию 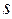 ,
, 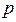 ,
, 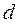 ,
, 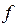 ,
, 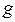 ,
, 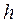 и т.д. Так, если электронные оболочки (энергетические подуровни) не полностью застроены эквивалентными электронами, то одной и той же электронной конфигурации атома может соответствовать несколько энергетических состояний (атомных термов), что является в свою очередь результатом взаимодействия электронов. Рассмотрим данный вопрос подробней. Уже не однократно подчёркивалось, что волновая функция электронной системы должна быть антисимметричной по отношению к перестановке любой пары частиц местами и учитывать как пространственные, так и спиновые координаты электрона. Поскольку электроны в атоме взаимодействуют между собой, то соответственно их векторы орбитальных и спиновых моментов должны складываться как векторные величины, давая вектора полного орбитального и спинового угловых моментов.
и т.д. Так, если электронные оболочки (энергетические подуровни) не полностью застроены эквивалентными электронами, то одной и той же электронной конфигурации атома может соответствовать несколько энергетических состояний (атомных термов), что является в свою очередь результатом взаимодействия электронов. Рассмотрим данный вопрос подробней. Уже не однократно подчёркивалось, что волновая функция электронной системы должна быть антисимметричной по отношению к перестановке любой пары частиц местами и учитывать как пространственные, так и спиновые координаты электрона. Поскольку электроны в атоме взаимодействуют между собой, то соответственно их векторы орбитальных и спиновых моментов должны складываться как векторные величины, давая вектора полного орбитального и спинового угловых моментов.
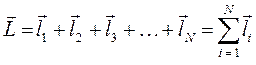
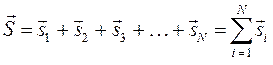
здесь:
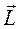 - орбитальный угловой момент атома;
- орбитальный угловой момент атома;
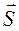 - спиновый угловой момент атома;
- спиновый угловой момент атома;
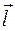 - орбитальный угловой момент отдельного электрона в атоме;
- орбитальный угловой момент отдельного электрона в атоме;
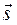 - спиновый угловой момент отдельного электрона в атоме.
- спиновый угловой момент отдельного электрона в атоме.
Физический смысл для случая многоэлектронных атомов, очевидно, будут иметь только орбитальный 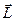 и спиновый
и спиновый 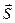 моменты атома, а не угловые моменты отдельных электронов
моменты атома, а не угловые моменты отдельных электронов 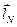 и
и 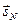 . В свою очередь орбитальные и спиновые моменты электронов в атоме, также взаимодействуют между собой. Результатом их сложения служит вектор полного (спин-орбитального) углового момента
. В свою очередь орбитальные и спиновые моменты электронов в атоме, также взаимодействуют между собой. Результатом их сложения служит вектор полного (спин-орбитального) углового момента 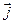 :
:
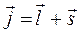
поскольку:
будем иметь соответственно:
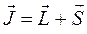
Учитывая выражения для векторов орбитального и спинового угловых моментов электрона:

а также, что:
будем иметь для вектора полного углового момента, выражение:
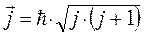
Очевидно проекции этих векторов на фиксированное направление в пространстве (ось 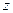 ), будут определяться выражениями вида:
), будут определяться выражениями вида:
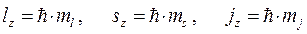
Распространяя полученные выражения на случай многоэлектронных систем, будем иметь соответственно:
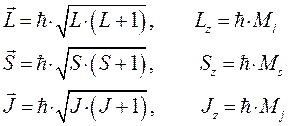
Каждое энергетическое состояние в атоме характеризуется определённым набором значений квантовых чисел углового момента 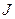 ,
, 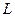 и
и 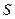 . Последние в свою очередь определяют полный, орбитальный и спиновый моменты атома. Проекции этих моментов на фиксированное направление в пространстве (в данном случае ось ) определяются квантовыми числами
. Последние в свою очередь определяют полный, орбитальный и спиновый моменты атома. Проекции этих моментов на фиксированное направление в пространстве (в данном случае ось ) определяются квантовыми числами 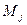 ,
, 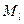 и
и 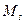 . Для описания энергетического состояния отдельного электрона в атоме, используют эквивалентные приведенным выше, квантовые числа углового момента
. Для описания энергетического состояния отдельного электрона в атоме, используют эквивалентные приведенным выше, квантовые числа углового момента 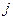 ,
, 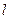 и
и 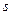 . Последние определяют полный, орбитальный и спиновый моменты электрона в атоме. Проекции этих моментов на фиксированное направление в пространстве (в данном случае ось ) определяются квантовыми числами
. Последние определяют полный, орбитальный и спиновый моменты электрона в атоме. Проекции этих моментов на фиксированное направление в пространстве (в данном случае ось ) определяются квантовыми числами 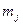 ,
, 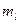 и
и 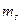 . Одной и той же конфигурации могут отвечать состояния с различными значениями и . Состояния эти сильно отличаются по энергии; в каждом из них векторы
. Одной и той же конфигурации могут отвечать состояния с различными значениями и . Состояния эти сильно отличаются по энергии; в каждом из них векторы 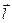 и
и 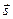 ориентированы по-разному, в результате чего межэлектронное отталкивание вносит различные вклады в общую электронную энергию атома. Последнюю принято выражать в единицах
ориентированы по-разному, в результате чего межэлектронное отталкивание вносит различные вклады в общую электронную энергию атома. Последнюю принято выражать в единицах 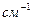 , а энергетические состояния с данными и называют атомными термами. Термин «терм» впервые появился в ранних работах по спектроскопии. При этом частоту каждой спектральной линии представляли в виде разности двух членов – термов
, а энергетические состояния с данными и называют атомными термами. Термин «терм» впервые появился в ранних работах по спектроскопии. При этом частоту каждой спектральной линии представляли в виде разности двух членов – термов 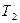 и
и 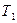 . В современной трактовке спектров, спектральная линия соответствует переходу атома из одного стационарного энергетического состояния (терма) в другое, а частота такого спектрального перехода, определяется выражением:
. В современной трактовке спектров, спектральная линия соответствует переходу атома из одного стационарного энергетического состояния (терма) в другое, а частота такого спектрального перехода, определяется выражением:
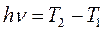
Каждый терм описывается набором квантовых чисел угловых моментов атома – орбитального, спинового и полного (спин-орбитального). Их определяют в рамках векторной модели атома, которая основана на рассмотрении векторного сложения угловых моментов электрона в атоме. Однако здесь сложение квантованных величин производится иначе, чем сложение соответствующих величин в классической механике. Так, в классической механике угловой момент определяется как аддитивная величина. Действительно, рассматривая вращательное движение некоторого твёрдого тела, можно заметить, что все точки такого тела будут двигаться по концентрическим окружностям, центры которых расположены на оси вращения. Каждая такая точка движется по окружности со своей линейной скоростью. Одинаковой для них, очевидно, будет являться только угловая скорость:
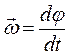
поскольку:
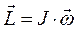
а также:
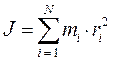
тогда соответственно будем иметь:
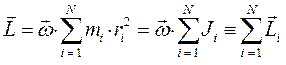
Таким образом, если у нас имеется система, состоящая из частиц с моментами 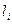 , тогда очевидно полный её момент будет представлять собой суперпозицию моментов , т.е.
, тогда очевидно полный её момент будет представлять собой суперпозицию моментов , т.е.
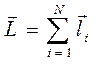
В квантовой механике сложение моментов означает нахождение возможных разрешённых значений для квантового числа результирующего вектора . В этом случае сложение производят с учётом пространственного квантования моментов. Так, если в качестве оси квантования выбрать ось, вдоль которой направлен первый вектор 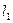 , тогда второй вектор
, тогда второй вектор 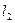 будет располагаться не под любыми углами, а в зависимости от того, какое значение имеет его проекция на выбранную ось. Рассмотрим способы нахождения квантовых чисел моментов и проекций моментов, а также величин моментов и проекций моментов на фиксированное направление в пространстве для результирующего вектора. Итак, пусть складываются следующие угловые моменты:
будет располагаться не под любыми углами, а в зависимости от того, какое значение имеет его проекция на выбранную ось. Рассмотрим способы нахождения квантовых чисел моментов и проекций моментов, а также величин моментов и проекций моментов на фиксированное направление в пространстве для результирующего вектора. Итак, пусть складываются следующие угловые моменты:
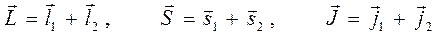
а также:
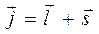
Для того чтобы найти все возможные значения квантовых чисел угловых моментов , и
Дата добавления: 2015-03-14; просмотров: 1689;