Хроматоґрафічні методи аналізу
У таких процесах розділення компонентів, як екстракція, у випадках, коли коефіцієнти розподілу між фазами для різних компонентів мало відрізняються один від одного, відокремити їх у одну ступінь не вдається. Щоб повторити розподіл багато разів, вдаються до динамічних методів – хроматоґрафії. У хроматоґрафічному процесі 2 фази – нерухома й рухома (що рухається відносно першої). Поверхня розподілу (межа) між цими фазами досить велика, що забезпечує інтенсивний перерозподіл речовин, які ми розділяємо, між фазами. Просуваючись, рухома фаза багато разів обмінюється з нерухомою цими речовинами, дозволяючи ефективно їх розділяти навіть при близьких коефіцієнтах розподілу.
Хроматоґрафію винайшов ботанік М. С. Цвєт, що у 1901 р. опублікував роботу про відокремлення забарвлених компонентів хлорофілу. Хроматоґрафічні процеси широко застосовують як у аналітичній хімії, так і у препаративному варіанті – щоб здобути продукти відокремлення.
Методи хроматоґрафії класифікують за різними ознаками:
1) За фізичною природою нерухомої та рухомої фаз – на рідинну (рухома фаза є рідкою) й газову хроматоґрафію. У свою чергу рідинну хроматоґрафію розділяють залежно від стану нерухомої фази на твердо-рідинну (нерухома фаза тверда) та рідинно-рідинну (нерухома фаза рідка). Рідинно-рідинну хроматоґрафію часто називають розподільчою хроматоґрафією. Газову хроматоґрафію розділяють на газоадсорбційну (нерухома фаза тверда) та газорідинну або газорозподільчу (нерухома фаза рідка).
2) За механізмом сорбції – на молекулярну й хемосорбційну. У молекулярній нерухома фаза та компоненти, що розділяють, взаємодіють за силами Ван-дер-Ваальса. До хемосорбційної відносять іонообмінну, осадову, ліґандообмінну, окисно-відновну. Сорбція тут спричиняється відповідними хімічними реакціями.
3) За засобом хроматоґрафування на фронтальну (аналізована суміш безперервно пропускають через нерухому фазу, й компоненти починають виходити з хроматоґрафічної колонки у порядку збільшення їх здатності до сорбції); проявну, або елюєнтну (де, увівши порцію суміші, через нерухому фазу надалі пропускають тільки рухому, вже без аналізованих компонентів); витіснювальну (де, щоб десорбувати компоненти, надалі пропускають рухому фазу, до якої додано витіснювач – речовину, що здатна сильно сорбуватись, вивільняючи аналізовані компоненти).
4) За формою нерухомої фази – на колоночну (нерухому фазу вміщують у колонку) та площинну (нерухома фаза – смуга паперу або тонкий шар сорбенту, що нанесений на скляну або металеву пластинку).
У сучасних приладах із колоночної елюєнтної хроматоґрафії на виході з колонки вміщують детектор. Він дає сиґнал, що залежить від концентрації компонентів, що визначають. На сиґнал мають впливати всі компоненти. Селективність детектора відносно окремих компонентів не має особливого значення, оскільки селективність аналізу забезпечується розділенням на колонці. Сиґнали детекторів забезпечують теплопровідність, електропровідність, поглинання випромінювань, теплота згоряння і т. ін.
В ідеалі хотілося б, щоб у елюєнтній хроматоґрафії окремі компоненти виходили з колонки як дуже вузькі зони. У дійсності зони розмиваються і компонент утворює так званий хроматоґрафічний пік – максимум концентрації (і відповідного сиґналу детектора), в околі якого сиґнал поступово сподає. Розмивання у максимумі концентрації спричиняється дифузією компонента як у напрямі руху розчинника, так і у перпендикулярному напрямі. Відіграє роль також і те, що рівновага процесів сорбції-десорбції встановлюється не миттєво, а потребує певного часу. Здатність колонки до утворення чіткого максимуму сиґналу на її виході описують у термінах моделі теоретичних тарілок. Цю величину використовують, щоб визначити ефективність хроматоґрафічного процесу, щоб порівнювати різні конструкції колонок, вибір рухомої та нерухомої фаз у проходженні (і розмиванні) зони, що відповідають тому чи іншому реаґентові.
Вводячи величину «число теоретичних тарілок», що характеризує якість розділення, розміри піку, тощо, використали модель й термінолоґію процесу розділення, що був відомий раніше за хроматоґрафію. Це процес ректифікації – ретельного відокремлення компонентів суміші летких компонентів через їх дистиляцію (переганяння). Одноразова дистиляція не дає бажаного розділення і потрібні повторні процеси. Їх можна орґанізувати у ректифікаційній колоні, де випари суміші компонентів, рухаючись уздовж колони, багаторазово конденсуються й знову випаровуються. Поступово випари все більше збагачуються більш летким компонентом (відповідно, конденсат – менш летким), поки не досягнуто бажаної якості розділення. Деякі конструкції колон містять так звані «тарілки», у яких міститься конденсат, через який пропускають випари, щоб досягти рівноваги. Чергова порція випарів, збагачена леткішим компонентом, піднімається з попередньої тарілки, що розташована нижче, у наступну, верхню тарілку. Надлишок конденсату, збагачений менш летким компонентом, зливається із верхньої тарілки у нижню (а із першої тарілки – назад, у «перегінний куб»). У теоретичній моделі обидві фази у тарілці вважають за повністю перемішані. Якість розділення визначається числом тарілок – чим їх більше, тим більше число послідовних дистиляцій імітує колона. Звичайно, що у хроматоґрафічному обладнанні таких «фізичних» тарілок не побачиш, тому у теоретичних моделях «тарілки» назвали «теоретичними».
Пов’язуючи число тарілок із формою хроматоґрами, використовують модель розмивання піку як випадкового процесу, що відповідає розподілові Ґаусса (або так званому нормальному розподілові). Ми маємо з цим розподілом ще зустрітися, вивчаючи розсіяння повторних вимірювань у задачах кількісного аналізу.
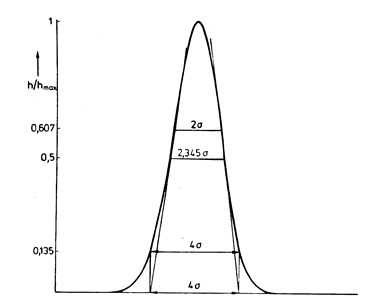
На рисунку зображено пік сиґналу на виході з колонки, форма якого − так звана крива Ґаусса. На осі абсцис може бути час, у який реєструють сиґнал, або (якщо розглядати пік, розподілений у колонці) відстань від початку колонки у певний момент часу. На осі ординат – концентрація компоненту в рухомій фазі (або сиґнал детектора, h, пропорційний концентрації. Як ординату відносну величину, h / hmax, де у знаменнику – максимальне значення сиґналу. Рівняння Ґауссова піка –
h = hmax × exp {‑ [(t – tR) / s ]2 / 2} = [А / (s 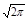 )] × exp {‑ [(t – tR) / s ]2 / 2},
)] × exp {‑ [(t – tR) / s ]2 / 2},
де tR – час, що відповідає максимумові сиґналу (величині hmax), А – загальна площа під ґрафіком, s − параметр розподілу, що зветься середнім квадратичним відхиленням. Ця функція симетрична відносно ординати, що відповідає tR . За властивостями розподілу Ґаусса, дотичні до функції проходять на відстані ± s від tR, й перетинають вісь абсцис на відстані ± »2 s від tR. Визначаючи s за даними експерименту, іноді вимірюють ширину кривої (різницю між значеннями t) для двох симетричних точок із сиґналом h / hmax = 0,5. Ця відстань дорівнює » 2,345 s. За моделлю число теоретичних тарілок дорівнює
n = (tR / s)2 » 5,54 (tR / w1/2)2 » 16 (tR / w)2,
де w1/2 – згадана вище ширина кривої для точок із h / hmax = 0,5, w – відстань між точками перетину дотичних із віссю абсцис, значення 5,54 » (2,345)2, 16 = (2 × 2)2.
|
Розрізняти зони від двох компонентів А та В можна, якщо відстань між ними більше за ширину обох зон. Щоб кількісно оцінити здатність розрізнити ці зони, уводять коефіцієнт розділення,
R s = (t R B – t R А) / (s A + s B) = 1,18 (t R B – t R А) / (w 1/2 A + w 1/2 B),
де останній індекс у величинах вказує, до якого компонента належить величина, а 1,18 = 2,345 / 2 – множник, що переводить w 1/2 у s. Тут вважається, що t R А < t R B (першим рухається компонент А).
Тонкошарова хроматоґрафія (ТШХ).ТШХ запропонована харківськими вченими М. А. Ізмайловим та М. С. Шрайбер, що у 1938 р. опублікували роботу з розділення алкалоїдів на пластинці з оксидом алюмінію. У ТШХ нерухому фазу наносять тонким шаром на скляну, металічну або пластмасову пластинку. Близько від краю пластинки, на так звану стартову лінію, наносять невелику кількість проби аналізованого розчину край пластинки занурюють у розчинник, що стає носієм для рухомої фази. Дією капілярних сил розчинник рухається уздовж шару сорбенту й із різною швидкістю переносить компоненти суміші, що спричиняє їх розділення у просторі.
Існують варіанти ТШХ, де рухому фазу пропускають (через ґніт) від середини пластинки до її країв, кільцями. Компоненти, що розділяють, рухаються за радіусами.
Розроблено також двовимірну ТШХ, де, пропустивши рухому фазу в одному напрямі, пластинку підсушують й пропускають рухому фазу іншого складу у перпендикулярному напрямкі.
| Основні характеристики ТШХ. На рисунку, поданому нижче, подано схему, що вказує розташування плям від різних компонентів на ТШХ. Він відповідає процесу ґрадуювання, коли краплі зразків для двох розчинів чистих компонентів було нанесено на стартову лінію a – a поруч одна від одної, але із зміщенням, щоб вони не змішувались, коли речовину переносить рухома фаза. Фронт її руху позначено лінією b‑b. Фронт перемістився від стартової лінії на відстань xf, середина плями від 1‑ї речовини – на відстань x1, а середина плями від 2‑ї речовини – на відстань x2. Рухаючись, плями розпливаються. Довжину плями від 1‑ї речовини на рисунку позначено як lн‑в, де індекси «н» та «в» відповідають нижній та верхній межі плями (так, відстань, яку пройшла нижню межа плями від 1‑ї речовини на рисунку позначено як xн). | 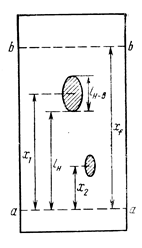
|
Відношення відстані від стартової лінії до середини плями речовини, до відстані, що проходить фронт рухомої фази, це величина Rf, характерна для речовини та складу рухомої та нерухомої фази. Для 1‑ї та 2‑ї речовин маємо
Rf1 = x1 / xf, Rf2 = x2 / xf .
У теоретичній моделі процесу величини Rf є постійними, якщо постійним лишаються коефіцієнти розподілу, а отже і частка часу, що проводить молекула речовини, що адсорбується (адсорбату) у рухомій фазі. Отже, слід виміряти величини Rf для стандартних зразків речовин, і використати їх, розшифровуючи хроматоґраму для аналізованої суміші.
За значеннями Rf ідентифікуємо речовини, здійснюючи якісний аналіз багатьох неогрґанічних та, особливо, органічних аналітів.
Якщо аналіт незабарвлений, то, висушивши пластинку з хроматоґрамою, діють на неї відповідними реаґентами, що дають інтенсивно забарвлені продукти із аналітами – проявляють хроматоґраму.
ТШХ приваблює низькою трудомісткістю, простотою та доступністю обладнання, універсальністю у застосуванні до різноманітних складних об’єктів. Здійснюючи точні виміри, можна оцінити ефективність процесу через число теоретичних тарілок та величину R s.
Порівнюючи площі плям (чи їх інтенсивність) для об’єкта та стандартів, здійснюють і кількісний аналіз. Із розвитком комп’ютерної техніки для кількісного аналізу у ТШХ розроблено відповідне проґрамне забезпечення. Хроматоґраму сканують, використовуючи серійні кольорові сканери з їх проґрамним забезпеченням, що дає 3 складових сприйняття кольору (відповідно трикольоровому зору й пов’язаних з ним конструкціям дисплеїв, принтерів, взагалі не тільки комп’ютерної, а й поліґрафічної техніки). Спеціальна комп’ютерна проґрама розшифровує зображення від сканера, виводячи з нього 3 складові залежності інтенсивності кольору від відстані між досліджуваною точкою хроматоґрами і стартовою лінією. Ці функції можна виводити як наочні ґрафіки (на екран чи на принтер), інтеґрувати, порівнювати одну з одною (наприклад, стандартного зразка та об’єкта), перераховувати у концентрації. Комп’ютерне обладнання досить поширене, доступне і недороге (порівняно з обладнанням для хроматоґрафічного аналізу іншими методами). Тому вказаний напрям є перспективним.
Як і в інших видах хроматоґрафії, на ефективність розділення впливає перш за все вибір нерухомої та рухомої фази. Досить зручно вживати пластинки з нерухомою фазою, що випускає промисловість. У нас визнання здобули пластинки марки «Сілуфол» на основі силікаґелю, що випускали у Чехословаччині.
Найбільш широке джерело оптимізації конкретної методики – вибір складу розчинника у рухомій фазі. Найчастіше використовують суміші орґанічних розчинників. Більш полярний компонент розчинника сприяє більшій сольватації полярних аналітів і зменшенню відповідних Rf. Вибір розчинника здійснюємо за порівняльними таблицями їх властивостей.
Умови ТШХ. Щоб сподіватись, що значення Rf у дослідах постійне, і цю величину можна використовувати як надійну характеристику у аналізі за ТШХ, слід підтримувати однакові умови як для стандартної речовини, так і для аналізованого об’єкта:
1) Зразки й об’єкт готують, використовуючи однаковий розчинник. Щоб зменшити витрати розчинника протягом руху фронту хроматоґрами й (у типовому випадку використання суміші розчинників) забезпечити постійний склад розчинника, процес здійснюють у спеціальній камері, атмосферу якої попередньо насичують випарами розчинника.
2) Концентрації аналізованих речовин та стандартів мають бути близькими, щоб залежності інтенсивності плями від концентрації були близькими до лінійних.
3) На пластинку наносять однакові об’єми розчинів об’єкта та стандартів. Площа плям на стартовій лінії має бути однаковою. Розчини наносять мікрокраплями й дають розчинникові випаритись після нанесення кожної краплі.
4) Розчини як об’єкта, так і стандартів готують, використовуючи той самий розчинник.
5) Зменшуючи похибки від коливання властивостей нерухомої фази, бажано розчини як об’єкта, так і стандартів наносити поруч.
Дата добавления: 2016-08-07; просмотров: 872;