Нарушения межуточного обмена аминокислот и их производных
Существуют наследственные и приобретённые нарушения интермедиарного обмена аминокислот. Вначале — об аномалиях, вызванных мутациями.
Рассматривая нарушения взаимопревращения аминокислот, отметим, что скорость их обмена в норме наиболее высока в нервной ткани. Поэтому разнообразные наследственные аминоацидопатии в психоневрологической практике известны, как одна из основных причин слабоумия.
В первую очередь, выделим наиболее частую и хорошо изученную мозаичную наследственную аминоацидопатию, имеющую несколько генокопий и частоту встречаемости 1/10000 (при частоте носительства — до 1/50 среди белых) — фенилпировиноградную олигофрению (см. также т.1 данного руководства, стр. 138). Данное расстройство аминокислотного обмена открыто в 1934 г. А. Феллингом, аутосомно-рецессивно и чаще всего встречается в скандинаво-балтийском регионе, а реже всего поражает африканцев и евреев.
Большинство случаев фенилкетонурии сопровождается дефицитом печеночного фермента фенилаланин-4-гидроксилазы. В полном соответствии с концепцией А. Гаррода это ведет к резкому увеличению концентрации фенилаланина в крови. Недостаток превращения фенилаланина в тирозин и подавление избытком фенилаланина активности тирозиназы поведет, в конечном итоге, к дефициту тирозиновых и триптофановых производных, включая пигмент меланин (что делает кожу, глаза и волосы больных светлыми), а также катехоламины (что проявляется гипотензией) и серотонин (что имеет отношение к развитию эпилептиформных «салаамовых судорог» и тремора).
Избыток фенилаланина метаболизируется обходными путями (рис. 13, отражающий основные пути метаболизма фенилаланина и тирозина), повышается концентрация его альтернативных продуктов метаболизма, избыток которых выводится с мочой. В крови и моче появляются в существенных количествах фенилпировиноградная и фенилмолочная кислоты, фенилацетилглутамин.
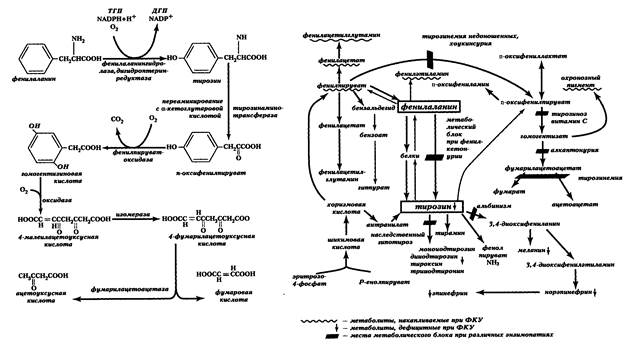
Рис. 13. Метаболизм тирозина и фенилаланина и его нарушения. ФКУ- фенилкетонурия
Скрининг-тест на гиперфенилаланинемию — реакция Феллинга (проба с полуторахлористым железом, дающая при положительном результате зеленое окрашивание) — выявляет в моче фенилпируват и широко внедрена в практику неонатологии во многих странах. Для диагностики используют и микробиологический тест Гатри, при котором кровь больного с гиперфенилаланинемией поддерживает рост ауксотрофного фенилаланинзависимого штамма бактерий (Л.Г. Смит, 1987).
При фенилкетонурии активируются минорные побочные пути метаболизма фенилаланина, при этом образуются метаболиты, практически отсутствующие в норме (фенилэтиламин, ортофенилуксусная кислота или фенилацетат — см. рис. 13). Эти соединения рассматриваются как нейротоксины и способны нарушать метаболизм липидов в мозге. В сочетании с дефицитом некоторых нейромедиаторов (например, серотонина) этот механизм считают ответственным за прогрессирующее снижение интеллекта у больных, выявляющееся через несколько месяцев после рождения.
Более 30 % из них имеют столь глубокую задержку психомоторного развития, что не ходят, а около 2/3 — не говорят. Слабоумие лишь у 4-5 % этих пациентов остаётся на уровне дебильности. У остальных коэффициент интеллекта не достигает 60 и регистрируется имбецильность или даже идиотизм (Дж. Джервис, 1954). Фенилкетонурии — одна из важнейших причин слабоумия у человека. Для больных характерны также наклонность к экземе и своеобразный мышиный запах мочи и пота, обусловленный фенилацетатом.
При классической форме уровень фенилаланина в крови больше 16 мг/дл (1 мМ/л). Основной метод лечения — ограничение потребления фенилаланина с целью снизить его концентрации в крови до уровня 3-12 мг/дл. Такое ограничение практикуется до полового созревания. Взрослые могут следовать менее строгой диете и потреблять фенилаланин.
Однако, в настоящее время доказано, что избыток фенилаланина и его минорные метаболиты тератогенны. Поэтому, если избыток фенилаланина не подвергается эффективному метаболизму, то у женщин, страдающих фенилкетонурией, он может оказывать пагубное действие на плод, вызывая множественные пороки развития. При беременности больные фенилкетонурией вынуждены возвращаться к бесфенилаланиновой диете (В. Кумар и соавт., 1997).
Болезнь вызвана генной мутацией в 12-й хромосоме и имеет несколько генокопий. Они связаны с различными мутантными аллелями фенилаланингидроксилазы, а также с дефектом другого фермента гидроксилирующей системы фенилаланина — дигидроптеридинредуктазы, необходимой для регенерации тетрагидроптерина, донора электронов для фенилаланин-4-гидроксилазной реакции.
Некоторые из мутантных аллелей фенил- аланин-4-гидроксилазы обладают пониженной, но выраженной активностью, и поэтому в их присутствии болезнь протекает как доброкачественная гиперфенилаланинемия — с небольшим повышением уровня фенилаланина в крови и без развития слабоумия. Форма, обусловленная дефектом дигидроптеринредуктазы, охватывает от 5 до 10% случаев болезни и, в отличие от классической, не лечится ограничением употребления фенилаланина. Она, таким образом, имеет худший прогноз (злокачественная гиперфенилаланинемия) и выявляется по снижению уровня фенилаланина крови после пищевой нагрузки тетрагидробиоптерином.
Алкаптонурия — аутосомно-рецессивная болезнь, на примере которой сэр Арчибальд Гаррод (1909) создал концепцию метаболического блока. Причина заболевания — дефект оксидазы промежуточного продукта распада фенилаланина и тирозина — гомогентизиновой кислоты. При крайне незначительной частоте (не более 1/1 000 000) болезнь наиболее часто наблюдается в Северной Ирландии (1/200000).
Алкаптонурия — яркая иллюстрация того, что генные мутации постоянно воспроизводят в человеческих популяциях дефектные аллели, несмотря на отбор, который, к тому же, не слишком эффективен в отношении болезней с таким поздним проявлением (после 30 лет). Болезнь описана еще у египетской мумии 3500-летней давности (Дж. Б. Вайнгарден, 1987). В норме фермент пара-оксифенилпируватдезоксигеназа, в присутствии витамина С, превращает в гомогентизиновую кислоту полученный из тирозина пара-оксифенилпируват. Затем, гомогентизиновая кислота должна в почках окислиться в присутствии Fe+2 и глутатиона до 4-малеилацетоуксусной кислоты. Если этот процесс тормозится, то накопление гомогентизиновой кислоты ведет к ее превращению полифенолоксидазой в хиноновые полифенолы, составляющие так называемый «охронозный пигмент», выводимый почками (рис. 13).
Это обусловливает потемнение мочи больных на воздухе и при подщелачивании. Хлорное железо при добавлении к моче больных окрашивается в голубой цвет. Пациенты, в буквальном смысле, выделяют с мочой... фотопроявитель. При попадании на фотобумагу подщелоченная моча больного вызывает ее почернение! Гомогентизиновая кислота ингибирует фермент лизилгидроксилазу, участвующий в синтезе коллагена, а охронозный пигмент (алкаптон) не полностью экскретируется с мочой, откладывается в основном веществе хрящей и других соединительно-тканных образований и делает их хрупкими, что со временем вызывает кальцификацию и дегенеративный артрит позвоночника, а также крупных суставов конечностей.
Первыми проявлениями болезни могут быть пигментация склер и хрящей ушных раковин (С.Я. Капланский, 1966). На вскрытии хрящи скелета, гортани, трахеи и другие соединительно-тканные образования у больных бывают тёмными и даже угольно-чёрными. Радикально болезнь не лечится. Степень остеохондропатии можно уменьшить, защищая активность лизилгидроксилазы большими дозами аскорбиновой кислоты.
Дата добавления: 2022-06-14; просмотров: 3168;