Лекция №8. Спектроскопия протонного магнитного резонанса
Основы метода. Область применения. Техника приготовления образцов. Чувствительность в экспериментах протонного магнитного резонанса. Растворители. Химический сдвиг. Спин-спиновое взаимодействие. Общие рекомендации по расшифровке спектров ПМР при структурном анализе.
Основы метода. Область применения
В соответствующих условиях в магнитном поле образец может поглощать электромагнитное излучение в радиочастотной области, причем частоты поглощения определяются свойствами образца. Поглощение обусловлено наличием ядер в молекуле. График в координатах частота поглощения – интенсивность сигнала представляет собой спектр ЯМР.
Все ядра несут заряд. В некоторых ядрах этот заряд «вращается» относительно оси ядра, и это вращение ядерного заряда генерирует магнитный диполь вдоль оси. Угловой момент вращающегося заряда может быть описан его спиновом числом I. Эти числа имеют значения 0, ½, 1, 3/2 и т.д. (I=0 означает отсутствие спина). Характеристическая величина генерируемого диполя выражается в величинах ядерного магнитного момента μ.
В спектрометрах стационарного типа (CW-типа) образец помещают в магнитном поле и облучают путем медленной развертки частоты в требуемом диапазоне частот. Спектр получают, записывая поглощение энергии как функцию частоты. В импульсном спектрометре образец помещают в магнитное поле и облучают мощным импульсом радиочастотной энергии, содержащим широкий диапазон частот, достаточный для облучения всего требуемого диапазона. Этот импульс возбуждает все ядра образца одновременно. Непосредственно вслед за импульсом ядра начинают вращаться в основное состояние и излучают поглощенную энергию. Детектор собирают эту энергию в виде сигнала свободной индукции (ССИ), который является суммой излучения всех ядер в зависимости от времени. Информация, содержащаяся в ССИ как функция времени, с помощью преобразования Фурье превращается в результирующий спектр, который является функцией частоты.
Спектры ПМР характеризуются двумя параметрами – химическим сдвигом и константами спин-спинового взаимодействия, которая коррелируется со структурой органического соединения и распределения электронной плотности в молекуле.
Интерпретация спектров ЯМР1Н основана на рассмотрение трех видов информации: на данных интегрирования, на значениях химических сдвигов отдельных сигналов и на величинах констант спин-спинового взаимодействия. Интегрирование сигналов лает соотношение протонов в отдельных группах. Величины химических сдвигов позволяют связывать сигнал с типом химического окружения. Важным является понимание эквивалентности отдельных протонов на основании данных по химическим сдвигам. Константы спин-спинового взаимодействия позволяют связать пары ядер друг с другом. Спин-спиновое взаимодействие позволяют определить типы соседних ядер.
Техника приготовления образцов.
Образец (в рутинных исследованиях – это раствор в дейтерированном растворителе в стеклянной ампуле с внешним диаметром 5 мм) помещают в датчик, который содержит передающую и приемную катушки и турбинку для вращения ампулы вокруг ее вертикальной оси, чтобы усреднить неоднородности поля.
Чувствительность в экспериментах протонного магнитного резонанса.
Чувствительность задается величиной отношения сигнала к шуму (S/N):
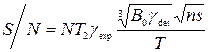 ,
,
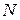 - число спинов в системе (концентрация образца),
- число спинов в системе (концентрация образца), 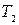 - время поперечной релаксации (определяет ширину линии),
- время поперечной релаксации (определяет ширину линии), 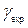 - гидромагнитное соотношение возбуждаемого ядра;
- гидромагнитное соотношение возбуждаемого ядра; 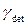 - гидромагнитное соотношение детектируемого ядра;
- гидромагнитное соотношение детектируемого ядра; 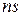 - число прохождений;
- число прохождений; 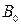 - внешнее магнитное поле;
- внешнее магнитное поле; 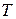 - температура образца.
- температура образца.
Обычный образец для регистрации протонного спектра ЯМР на спектрометре частотой 300 МГц содержит около 10 мг вещества примерно в 0,5 мл растворителя в стеклянной ампуле с внешним диаметром 5 мм. Существуют микродатчики, в которые можно помещать ампулы с внешним диаметром 1,0; 2,5 и 3 мм, и они обеспечивают более высокую чувствительность. При благоприятных условиях в микроампуле 1 мм (объемом 5 мкл) можно получить спектр от 100 нг соединения средней молекулярной массы на спектрометре с частотой 600 Мгц.
Создание криогенных датчиков существенно уменьшило количество образца, требуемое для записи спектра ЯМР1Н. Тепловой шум датчика и электроники первой ступени усилителя является преобладающим шумом в экспериментах. Очевидно, что спектрометры с наиболее сильными полями дают наилучшую чувствительность.
При ограниченном количестве образца обычно наилучшим вариантом является использование наиболее сильных из возможных полей и криодатчик с наименьшим диаметром. Криокапиллярный проточный микродатчик, который позволяет работать с несколькими нанограммами материала в 1 мкл растворителя, обладает наивысшей чувствительностью образцов, доступных в ограниченных количествах.
Растворители
Идеальный растворитель не должен содержать протонов. Кроме того, желательно, чтобы растворитель был инертным, низкокипящим и недорогим. Для современных приборов необходимы дейтрированные растворители, поскольку стабилизация поля магнита осуществляется с помощью дейтеририя растворителя. Прибор имеет дейтериевый «канал», который постоянно изменяет и подстраивает поле к частоте дейтерированного растворителя.
Дейтерохлороформ (CDCl3) используется в большинстве случаев. Маленький узкий протонный сигнал при 7,26 м.д. от примесного хлороформа (CHCl3) мешает в редких случаях. Для очень разбавленных образцов доступен CDCl3 100 %-ной изотопной чистоты.
Наиболее доступные коммерческие растворители – это ацетон-d6 (2,04 м.д.), бензол- d6 (7,17 м.д.), дихлорметан- d2 (3,53 м.д.), диэтиловый эфир- d10 (3,34 м.д.), 1,2-дихлорэтан- d4 (3,72 м.д.) и т.д.
Много неприятностей могут доставить следы обычных лабораторных растворителей: вода, ацетон, ацетонитрил, диоксан, матанол, пиридин и т.д.
Химический сдвиг
Разность положения сигнала данного протона и положения сигнала протона стандарта называется химическим сдвигом данного протона. Протоны в «разном» химическом окружении имеют различные химические сдвиги. И наоборот, протоны в «одинаковом» химическом окружении имеют один и тот же химический сдвиг. Химический сдвиг является критерием структурной близости ядер в молекуле. При этом ядра с одинаковыми химическим сдвигом называются химически эквивалентными. Таблицы химических сдвигов могут использоваться для отнесения сигналов в спектрах ЯМР и определения структуры молекул.
В качестве эталона для измерения химических сдвигов протонов чаще всего используют тетраметилсилан (ТМС, Si(CH3)4), содержащий 12 структурно эквивалентных сильно экранированных протонов, образующих легко опознаваемый узкий сигнал в сильнопольной (правой) части спектра. ТМС – химически инертное вещество, летучее, растворимо в большинстве органических растворителей, его молекула симметрична. Это соединение дает интенсивный, узкий, синглетный сигнал в спектре ЯМР1Н, и его протоны «экранированы» в большей степени, чем протоны почти всех органических соединений.
Сигналы стандарта ТМС (тетраметилсилан, Si(CH3)4) помещают на правом краю спектра и принимают его за нуль шкалы либо в герцах, либо в δ-шкале. Химические сдвиги можно сообщать в безразмерных единицах (обозначаемых δ или м.д.). Химические сдвиги не зависят от рабочей частоты используемого спектрометра. Это достигается делением химического сдвига, выраженного в герцах, на рабочую частоту спектрометра, выраженную мегагерцах.
Для растяжки химических сдвигов следует использовать максимально сильные доступные магнитные поля. Для объяснения закономерностей химических сдвигов весьма плодотворной является концепция электроотрицательности заместителей. Это концепция говорит о том, что электронная плотность вокруг протонов в метильных группах ТМС высока, и эти протоны сильно экранированы.
Спин-спиновое взаимодействие.
Это явление может быть описано как взаимодействие спинов протонов через электроны связи. Согласно принципу Паули, электроны, связывающие два ядра, спарены, т.е. их спины антипареллельны. В магнитном поле имеется определенная тенденция для каждого из ядер спаривать свой спин со спином одного из связывающих электронов таким образом, чтобы большинства из них были антипараллельными; это соответствует устойчивому состоянию. Обычно спин-спиновое взаимодействие распространяется очень слабо далее трех связей, если только это не напряженные циклы (такие как малые циклы), мостиковые системы, делокализованные системы связей (в ароматических или ненасыщенных структурах) или система из четырех связей, находящихся в конфигурации W. Спин-спиновое взаимодействие через две связи называется геминальным, через три связи – вицинальным:
| Н – С – Н | Н – С – С – Н |
| Геминальное спин-спиновое взаимодействие через 2 связи (2J) | Вицинальное спин-спиновое взаимодействие через 2 связи (3J) |
Разность частот в герцах между компонентами в дублете пропорциональная эффективности взаимодействия и характеризуется константой спин-спинового взаимодействия J которая не зависит от величины внешнего магнитного поля . В то время как химические сдвиги обычно покрывают интервал 3600 Гц при рабочей частоте прибора 300 МГц, величины констант спин-спинового взаимодействия редко превышают 20 Гц.
Пока разность химических сдвигов в герцах (Δν) гораздо больше, чем константа спин-спинового взаимодействия (произвольно можно принять, что Δν/J), в спектре проявляются простые сигналы в виде двух дублетов. По мере того как отношение Δν/J становится меньше, дублеты приближаются друг к другу, интенсивность двух внутренних линий возрастает, а двух внешних линий – уменьшается. Положение химического сдвига для каждого из протонов не будет соответствовать центру дублета, а находится в «центре тяжести» и определяет без расчета с невысокой точностью.
В простых мультиплетах первого порядка число линий определяется числом взаимодействий с соседними протонами, имеющими одни и те же (или очень слабо различающиеся) константы спин-спинового взаимодействия. Соседними являются геминальные или вициальные протоны, т.е. протоны, отстающие на две или три связи; дальние константы спин-спинового взаимодействия обычно гораздо меньше по величине. Один протон вызывает расщепление сигнала в дублет, два протона с одинаковым спин-спиновым взаимодействием приводят к триплету. Отсюда мультиплетность равна n+1, где n – число соседних протонов, имеющих одинаковые константы спин-спинового взаимодействия с данным протоном. Общая формула, справедливая для подсчета линий при взаимодействии с n ядрами со спинами I , записывается в виде 2nI+1, где I – спин ядра. Относительные интенсивности линий в простых мультиплетах первого порядка также зависят от n. В дублете (n =1) это соотношение равно 1:1, в триплете (n =2) – 1:2:1, в квартете (n =3) – 1:3:3:1 и т.д.
Требования, которые должны быть выполнены, чтобы наблюдался простой мультиплет первого порядка, можно суммировать следующим образом:
- отношение Δν/J должно быть больше примерно 8; Δν – расстояние в герцах между средними точками мультиплетов, связанных спин-спиновым взаимодействием; J – константа спин-спинового взаимодействия.
- число линий в мультиплете равном n+1, где n – число соседних протонов, имеющих одинаковые константы спин-спинового взаимодействия с данным протоном.
- расстояние в герцах между линиями простого мультиплета первого порядка равны константам спин-спинового взаимодействия.
- простые мультиплеты первого порядка симметричны относительно центра, и наиболее интенсивная линия (линии) находится в центре.
Общие рекомендации по расшифровке спектров ПМР при структурном анализе
Приступая в интерпретации спектра ПМР для определения строения чистого вещества, следует прежде всего определить число присутствующих в молекуле структурных типов протонов и относительное число протонов каждого типа. Это достигается выявлением в спектре отдельных сигналов (синглетных и сложных) и оценки их относительной интенсивности по ступенькам интегральной кривой (а при отсутствии интегратора – геометрическим и весовым способом определения площадей). Абсолютное число протонов каждого типа находят по их относительному числу с учетом брутто-формулы или иной неспектральной информации об исследуемом веществе. Если нет таких дополнительных сведений, то в качестве исходного предположения принимают, что самый слабый сигнал в спектре соответствуют одному протону или, точнее, тому минимальному числу протонов, которое согласуется с целочисленностью протонов других групп. В ходе последующего анализа данных это предварительное допущение может быть пересмотрено и учтены возможности наличия вдвое, втрое, или еще большего числа эквивалентных протонов.
Выявление сложной структуры сигналов является важной и ответственной расшифровки спектра ЯМР. Дублетный сигнал иногда трудно отличить от двух близко расположенных синглетов одинаковой интенсивности. При этом надо учитывать, что возникновение дублета может быть следствием спин-спинового взаимодействия не только с соседним протоном, но и с магнитным ядром другого элемента, имеющим спин ½ (фтор). При возникновении дублета от спин-спинового взаимодействия с протоном сигнал последнего будет также расщеплен (с тем же расстоянием между линиями тонкой структуры), и он должен быть обязательно опознан для подтверждения дублетной природы близко расположенных линий.
В случае затруднений в истолковании некоторых участков спектра можно рекомендовать обозначить их как «мультиплеты» или «перекрывающиеся мультиплеты» и приступить ко второму этапу расшифровки спектра.
Второй этап состоит в определении положения в молекуле магнитных ядер, дающих обнаруженные в спектре сигналы. Основанием для такого соотнесения сигналов служат их положение (химический сдвиг) и структура (мультиплетность, значения констант спин-спинового взаимодействия). Прежде чем сопоставлять химический сдвиг каждого из сигналов с табличными данными, полезно учесть перечисленные ниже общие закономерности расположения сигналов в спектрах ПМР.
1. В сильнопольной (правой) части спектра с δ<2 м.д. располагаются сигналы относительно хорошо экранированных протонов алкильных и циклоалкильных радикалов, не находящихся по соседству с электроотрицательными заместителями, кратными связями и ароматическими кольцами. Здесь же находятся сигналы протонов связей Si-H и Р-Н. Удобным репером могут служить в большинстве случаев интенсивные сигналы метильных групп парафиновых цепей с δ около 1 м.д.
2. На противоположном (левом) слабопольном конце спектра (обычно регистрируемого в интервале δ от 0 до 10 м.д.) наблюдаются сигналы сильно разэкранированных альдегидных протонов (δ 9 – 10 м.д.) и протонов различных ароматических колец с δ от 6 до 9 м.д. (чаще всего 7 – 8 м.д.). Сдвиг 6 – 7 м.д. могут иметь также «олефиновые» протоны (при связях С=С), подверженные дополнительным эффектам дезэкранирования соседними электроотрицательными группами или ароматическими кольцами. Сигналы протонов при sp3 – углеродных атомах попадают в эту слабопольную область, только если они сильно разэкранированы совместным действием нескольких соседних явно электроотрицательных заместителей (хлороформ, бромоформ, динитрометан и т.п.).
3. В средней части спектра (δ от 2 до 6 м.д.) располагаются сигналы «олефиновых» и «ацетиленовых» протонов, а также протонов алкильных и циклоалкильных радикалов, дезэкранированных соседством электроотрицательных групп, двойных связей или ароматических колец.
4. Особую группу составляют сигналы протонов, связанных с гетероатомов (O, N, S), участвующих в процессах межмолекулярного обмена, образования водородных связей и по этим причинам не имеющих строго фиксированных значений химических сдвигов. Положение этих сигналов может существенно изменяться в зависимости от природы растворителя , концентрации и природы самого соединения. В общем, сигналы протонов групп ОН, NH, SH алифатических и алициклических спиртов, аминов и меркаптанов располагаются в правой части спектра ПМР (δ<5 м.д.). Если же эти группы связаны с sp2-нибридным атомом углерода (в енолах, карбоновых кислотах, фенолах и амидах), то протоны сильно разэкранируются и их сигналы оказываются в левой части спектра (δ>5 м.д.). Неопределенность положения сигналов гидроксильных и аминогрупп не очень осложняет их отнесение, так как именно вследствие межмолекулярного обмена эти сигналы имеют заметно большую ширину и опознаются по характерному контуру.
После отнесения сигналов на основании химических сдвигов следует определить число неэквивалентных геминальных и вацинальных протонов, находящихся в спин-спиновом взаимодействии с каждой из выявленных групп протонов. Это определение, производится по структуре сигналов, учитывая, что в спектрах первого порядка взаимодействие с n протонами приводит к расщеплению сигнала при n+1 линий. Так, дублетный сигнал (n+1 =2) указывает, что на расстоянии двух-трех ковалетных связей от протона или протонов, дающих этот дублет, находится один структурно неэквивалентный протон. Триплетный сигнал (n+1 =3) свидетельствует о наличии по соседству двух протонов и т.д.
По окончании второго этапа расшифровки спектра ПМР исследователь располагает некоторым числом выявленных по спектру структурных фрагментов (водородсодержащих радикалов). Задача последующего (завершающего) этапа определения структуры состоит в компоновке этих фрагментов таким образом, чтобы их свободные валентности оказались насыщенными, а окончательная структура полностью соответствовала всей совокупности сведений об исследуемом веществе. После исчерпывающего анализа спектра ПМР по всем параметрам может остаться некоторая неопределенность в установлении структурной формулы. Для устранения этой неопределенности, как и при использовании других физических методов, требуется привлечение дополнительной информации, прежде всего брутто-формулы.
Лекция №9. Спектроскометрическая идентификация органических соединений (совместное использование УФ-, ИК-, ЯМР-, ПМР- и масс-спектроскопии)
Структурный анализ соединений обычно включает четыре этапа:
1. Как правило, необходимой предпосылкой определения строения молекулы является установление числа и природы образующих ее атомов, т.е. брутто-формулы. Брутто-формула может быть установлена химическими методами элементного анализа и определения молекулярной масс. Из всех физических методов для этой цели наиболее пригодна масс-спектрометрия.
2. Выявление в молекуле определенных атомных группировок (функциональных групп и фрагментов углеродного скелета). Таким образом, осуществляется отнесение исследуемого вещества к той или иной группе (классу) органических соединений (классификация или групповая идентификация). В зависимости от возможностей метода и природы исследуемого объекта групповая идентификация осуществляется на разных уровнях:
а) отнесение к классу веществ с очень общей и неполной характеристикой структуры (циклоалкан, олефин, спирт, простой эфир, амин и т.д.);
б) определение принадлежности к тому или иному гомологическому ряду (например, ряд бензола, предельных спиртов, ацетиленовых спиртов, виниловых эфиров и т.д.);
в) установление групп изомеров с более или менее определенными особенностями структуры.
3. Вывод структурной формулы путем сопоставления состава, данных предварительной классификации и компоновки выявленных фрагментов структуры, включая все участки углеродного скелета.
4. Получение основных сведений о конфигурации и конформации молекул.
Простейшим способом доказательства его структуры может служить отождествление (идентификация) с соединением известной структуры.
Ни один из существующих методов структурного анализа не может считаться универсальным и достаточным для полного установления строения молекул. Даже при использовании наиболее эффективных методов (например, ЯМР) приходится привлекать дополнительную прямую или косвенную информацию о качественном и количественном составах вещества и некоторых деталях его структуры. Источником такой информации могут быть самые разнообразные физические и химические исследования, которые следует разумно сочетать для наиболее целесообразного и простого решения поставленной задачи.
Выбор оптимальных комбинаций различных методов зависит, конечно, от самого объекта исследования (первичной информации о его природе) и реальных возможностей данной лаборатории. Если одинаково доступны все основные современные методы исследования, то наиболее универсальным следует считать сочетание масс-спектроскопии, инфракрасной спектроскопии и ЯМР. Последовательность их использованиине имеет принципиального значения, но обычно оказывается целесообразным начинать с технически более простых и доступных методов(ИК- и УФ-спектры, рефрактометрия), а затем переходить к более сложным (ЯМР, масс-спектроскопия) и, наконец, привлекать в случае необходимости более специальную технику (измерение моментов диполя и др.). Поскольку обязательных общих рецептов совместной интерпретации физических данных не существует, типичный ход рассуждений рассмотрим на примере нескольких подробно разобранных конкретных задач. При этом следует обратить внимание не на отдельные факты и числовые данные, а на логику и последовательность заключений, взаимодействие и взаимодополнение разных методов, а также на использование дополнительной (неспектральной) информации. Предлагаемый ход решений отнюдь не является единственно возможным, и весьма полезно повторно разобрать примеры с иной последовательностью трактовки исходных данных.
При спектрометрическом изучении органического соединения обычно применяется следующая стратегия. Сначала масс-спектрометрически определяют его элементарный состав (молекулярную формулу) и предварительно отмечают характер его фрагментации. Затем с помощью инфракрасной спектроскопии определяют природу функциональных групп, а электронный спектр позволяет выяснить, сопряжены ли эти функциональные группы или нет. Далее по данным спектроскопии ЯМР1Н и 13С, анализируя окружении атомов водорода и углерода в изучаемой молекуле, выбирают одну или несколько наиболее вероятных структур. Наконец, полученные каждым методом данные сопоставляют друг с другом и перепроверяют с тем, чтобы убедиться, что они не противоречат найденному решению задач.
Часто задачу выяснения строения можно решить путем удачного подбора только одного или двух из указанных спектроскопических методов, иногда подтвердив полученные данные простым химическим тестом или посредством определения температуры плавления или кипения. В более сложных случаях необходима полная информация, и тогда необходимо постоянно проверять соответствие друг другу данных, получаемых с помощью различных методов. Подобный сравнительный критический анализ данных – это ключ к успешному решению задачи.
С практической точки зрения важно обращать внимание на условия регистрации и особенности интерпретации спектров. Существенным моментом, является вычитание из спектра пиков, отвечающих растворителю, или намеренное расширение шкалы при записи сигналов вне обычного рабочего диапазона прибора.
Полученные с помощью четырех основных методов: электронной и инфракрасной спектроскопии, масс-спектрокопии и спектроскопии ядерного магнитного резонанса данные обычно позволяют предложить для неизвестного соединения по меньшей мере ковалентную структуру, а часто дают возможность сделать определенные выводы и о его относительной стереохимии.
Список литературы, использованной при составлении лекций
1. Сильверстейн Р. Спектрометрическая идентификация органических соединений [Текст] / Р. Сильверстей, Ф. Вебстер, Д. Кимл. – М.: БИНОМ. Лаборатория знаний, 2012. – 557 с.
2. Воловенко Ю.М. Спектроскопия ядерного магнитного резонанса для химиков. Учебник для химических специальностей вузов [Текст] / Ю.М. Воловенко, В.Г. Карцев, И.В. Комаров, А.В. Турова, В.П. Хиля. – М.: Издано Международных благотворительным фондом "Научное Партнерство", 2011. – 704 с.
3. Преч Э. Определение строения органических соединений. Таблицы спектральных данных [Текст] / Э. Преч, Ф. Бюльманн, К. Аффольтер. – М.: Мир; БИНОМ. Лаборатория знаний, 2006. – 438 с.
4. Браун Д. Спектроскопия органических веществ [Текст] / Д. Браун, А. Флойд, М. Сейнзбери. – М.: Мир, 1992. – 300 с.
5. Идентификация органических соединений [Текст] / Под ред. Р. Шрайнер, Р. Фьюзон, Д. Кертин и др. – М.: Мир, 1983. – 703 с.
6. Казицына Л.А. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии [Текст] / Л.А. Казицынв, Н.Б. Куплетская. Учеб. пособие для вузов. - М., "Высшая школа", 1971. – 264 с.
7. Иоффе Б.В. Физические методы определения строения органических соединений [Текст] / Б.В. Иоффе, Р.Р. Костиков, В.В. Разин. Учеб. пособие для химических вузов. Под ред. Иоффе Б.В. – М.: Высш. шк., 1984. – 336 с.
Дата добавления: 2017-06-02; просмотров: 493;